

本文来自微信公众号“兴证医药健康”,作者:兴业证券医药小组。
摘要
中和抗体具有预防与治疗的双重作用,有望成为卫生事件的“特效药”:中和抗体是当病原微生物侵入人体时B淋巴细胞产生的一类抗体,能够与病毒表面S蛋白结合,阻断病毒入侵细胞。相比小分子药物和血浆疗法,中和抗体特异性和安全性更好,有望成为“特效药”。
关于中和抗体的开发策略的几点思考:1)临床前研发:针对卫生事件的病毒中和抗体药物研发的靶点集中在S蛋白的受体结合域RBD;候选中和抗体药物中绝大多数是从康复病人的血液里用单B细胞克隆技术筛选出来的,也有再生元等少数公司用到了转基因小鼠平台;中和抗体的临床前研发难度不大,重点是防止因病毒突变导致中和抗体药物失效,方法包括采用抗体鸡尾酒、选取不易发生突变的靶点等。2)临床研发:中和抗体的临床试验也是分3期,III期临床试验会针对多个适应症进行,包括高风险感染者的预防、轻至中度患者治疗和重症住院患者治疗等。中和抗体研发的关键在于临床试验的速度和效率。3)规模化生产:中和抗体的规模化生产能力也很重要,临床试验进展较快的企业已纷纷开展生产合作,以期在较短时间内实现中和抗体药物的大批量供应,如再生元(REGN.US)×罗氏、礼来(LLY.US)×安进(AMGN.US)、Vir Biotechnology(VIR.US)×Biogen(BIIB.US)×三星生物×药明生物(02359)等。
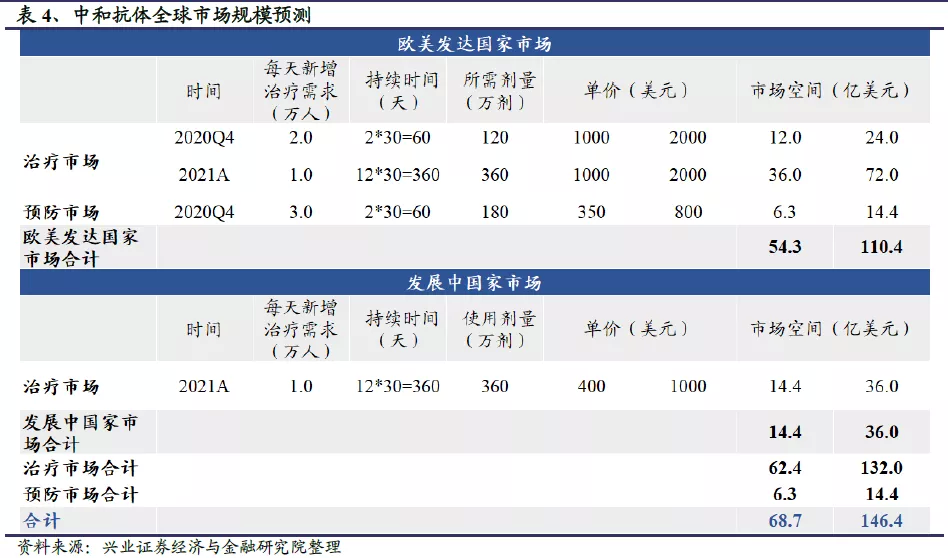
百亿美元级蓝海市场,中和抗体商业化价值大:假设公共卫生事件持续到2021年末,我们预计中和抗体的商业化市场空间可达69-146亿美元。其中欧美发达国家市场空间更大,达到54-110亿美元;发展中国家市场规模约14-36亿美元。另一方面,中和抗体用于患者治疗的市场空间更大,约62-132亿美元;用于高风险人群预防时,因为价格高、预防时间短等劣势,疫苗研发成功后很容易被替代,市场规模相对较小,或将不超过10亿美元。中和抗体的利润空间广阔,且赛道玩家相比疫苗更少。
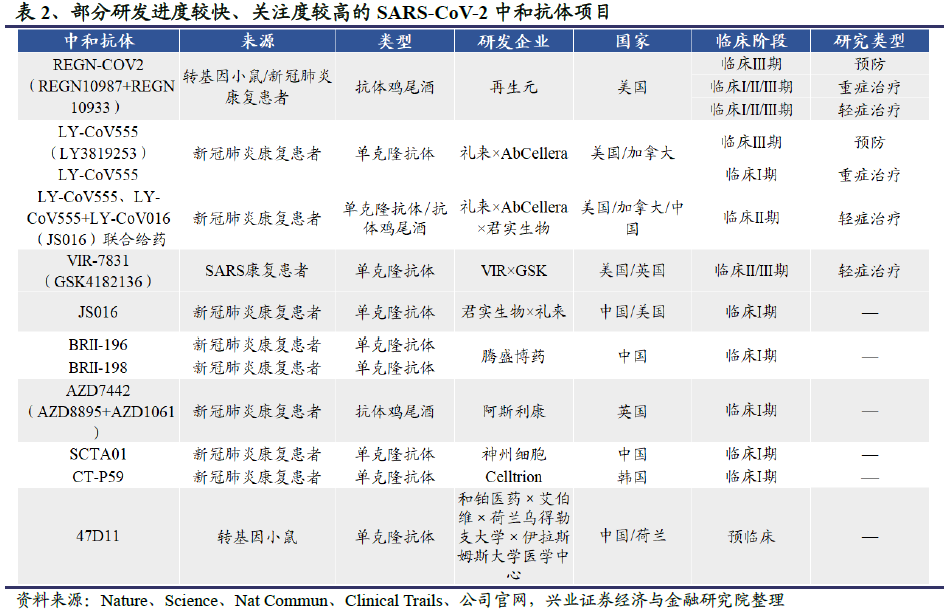
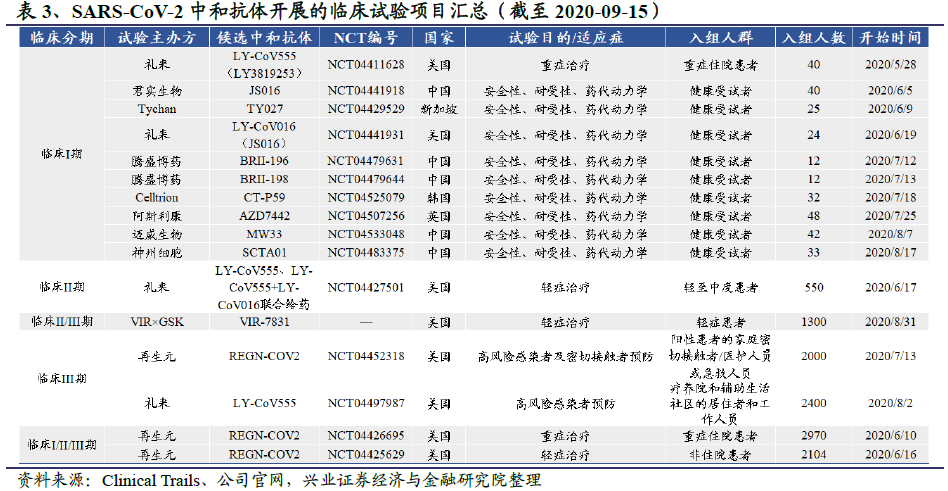
中和抗体的临床研发进展:目前全球共有12个中和抗体项目进入临床,其中进展较快的中和抗体研发项目包括由再生元(Regeneron)公司开发的中和抗体鸡尾酒疗法REGN-COV2,礼来和加拿大AbCellera公司联合开发的中和抗体LY-CoV555,Vir Biotechnology和GSK联合开发的VIR-7831。REGN-COV2和LY-CoV555已进入以预防为目的的III期临床试验阶段,三者均已进入以治疗为目的的II/III期临床试验阶段。国内公司中,君实生物(01877)和中科院微生物所合作开发的JS016领跑,成为中国第一个、全球第二个进入临床试验的病毒中和抗体疗法。
风险提示
公共卫生事件提前结束或疫苗先行研发成功的风险、中和抗体研发失败的风险、竞争格局加剧的风险。
报告正文
1、公共卫生事件持续肆虐,急需“特效药”救黎民于水火
1.1、全球公共卫生事件现状
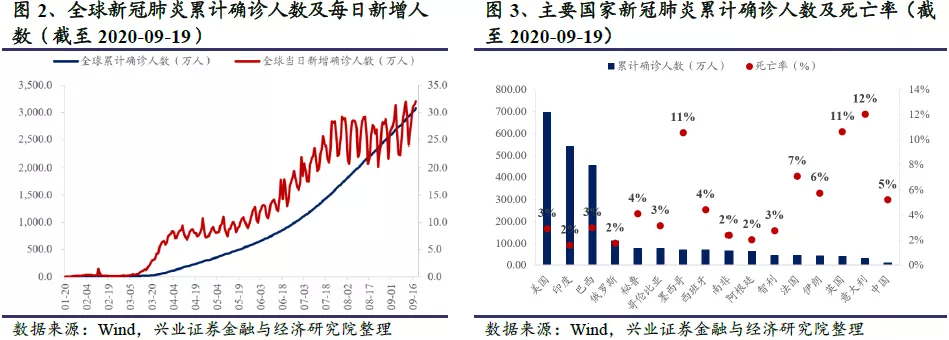
2019年12月以来,公共卫生事件持续肆虐,对全球的社会生活和经济发展造成了极大的冲击和影响。目前包括我国在内的部分国家公共卫生事件已经得到控制,但仍时有反复;而在全球更多国家,公共卫生事件仍在恶化,确诊病例和死亡病例数持续攀升。截至2020年9月20日,全球累计确诊人数超过3000万人,累计死亡人数超96万人,全球平均死亡率约为3.1%。美国累计确诊人数接近700万人,死亡人数超过20万人,居全球首位;印度紧随其后,累计确诊人数超过500万人,每日新增确诊人数不断创下新高,大有赶超美国的势头。
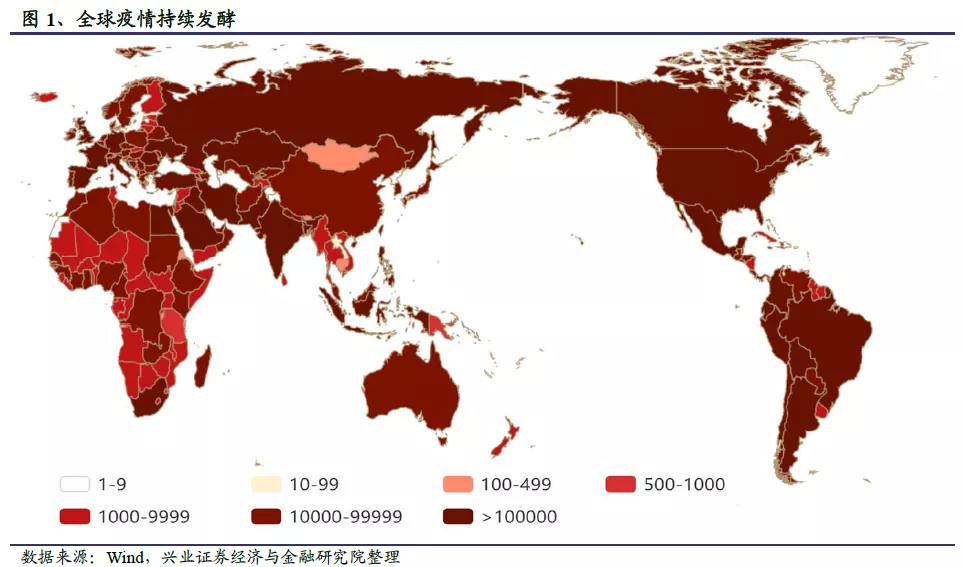
中国等公共卫生事件蔓延及控制较早的国家死亡率普遍在5%左右;英国、法国、意大利等欧洲国家因3月公共卫生事件进展迅速,救治不及时导致死亡率高达10%左右;美国、巴西、印度等国家因为感染人口基数过大,单看死亡率并不高,但死亡人数均达到几万甚至几十万量级。2020年以来公共卫生事件一波未平一波又起,全球遍地开花,随着公共卫生事件的持续发酵,死亡人数仍将不断攀升。
1.2、公共卫生事件是近代流行病史上规模最大的流行病之一
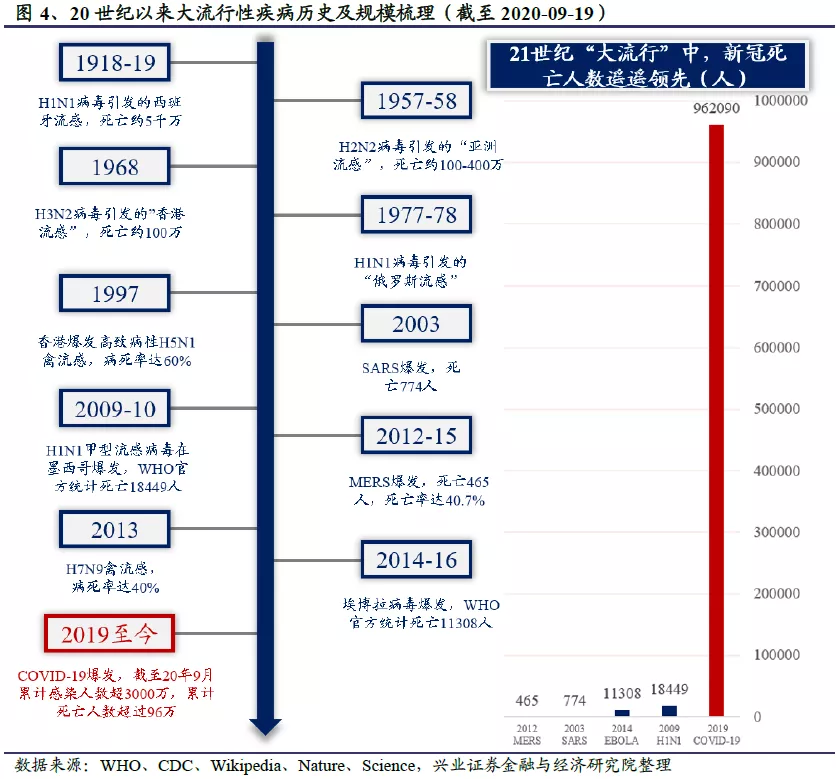
卫生事件已经成为近代流行病史上最大规模的流行病之一,公共卫生事件的持续肆虐和大规模扩散给全球公共卫生领域带来了极大挑战。截至2020年9月19日,COVID-19导致全球超过96万人死亡,是进入21世纪以来最大规模的流行病,造成的死亡人数远超2019年爆发的H1N1甲型流感、2014年爆发的埃博拉(EBOLA)病毒病和2003年令国民“闻风丧胆”的非典(SARS病毒)。即使放眼到整个近现代史(1840年至今),卫生事件导致的死亡人数也仅次于H1N1,历史上H1N1甲型流感卷土重来制造过多次“大流感”,但进入21世纪以后,还未曾引发过导致上百万人死亡的“大灾难”。
2、病毒病原学与现有治疗方案
2.1、病原学特征—与SARS-CoV同属β-冠状病毒,但传染性更强
国际病毒分类学委员会将该病毒命名为 SARS-CoV-2,世界卫生组织(WHO)将它引起的疾病被命名为 “COVID-19”。从名字也可以看出,病毒是 2002 年出现的严重急性呼吸综合征冠状病毒(SARS-CoV) 的“姐妹”。实际上SARS-CoV-2、SARS-CoV和中东呼吸综合征病毒(MERS-CoV)都属于β-冠状病毒,其中SARS-CoV-2是一种有包膜结构的单股正链RNA病毒,颗粒呈圆形或椭圆形,直径约60-220 nm。其基因特征与SARS-CoV和MERS-CoV具有一定同源性,但亦存在明显区别,例如传染性更强。
传播性:冠状病毒在人与人之间通常通过咳嗽和打喷嚏、与感染者密切接触或接触已感染的表面进行传播,其次通过嘴、鼻子或眼睛传播。
敏感性:该病毒SARS-CoV-2对热敏感,56 °C下加热30分钟、乙醚、75%乙醇、含氯消毒剂、过氧乙酸和氯仿等脂溶剂均可有效灭活病毒。
2.2、公共卫生事件的病毒S蛋白是疫苗和抗体药物研发的主要靶点
公共卫生事件的病毒具有包膜结构,有4种主要的结构蛋白:其中3种在膜上,分别是膜糖蛋白(Membrane Protein,M)、小包膜蛋白(Envelope Protein,E)和刺突糖蛋(Spike Protein,S);还有1种是与病毒RNA结合的核衣壳蛋白(Nucleocapsid,N)。少数种类还有血凝素糖蛋白(Haemaglutinin-esterase,HE)。S蛋白能够识别并结合宿主细胞表面受体,在介导病毒包膜与细胞膜融合方面有关键的作用;M蛋白参与病毒包膜的形成与出芽过程;N蛋白与病毒RNA结合,是一种具有高度免疫原性的蛋白,参与基因组复制和细胞信号通路的调节。由于N蛋白的序列保守性和强大的免疫原性,常被用于开发冠状病毒诊断检测试剂。HE蛋白则是构成包膜的短凸起,可能与冠状病毒早期吸附有关,某些冠状病毒的HE蛋白可引起红细胞凝集及红细胞吸附。
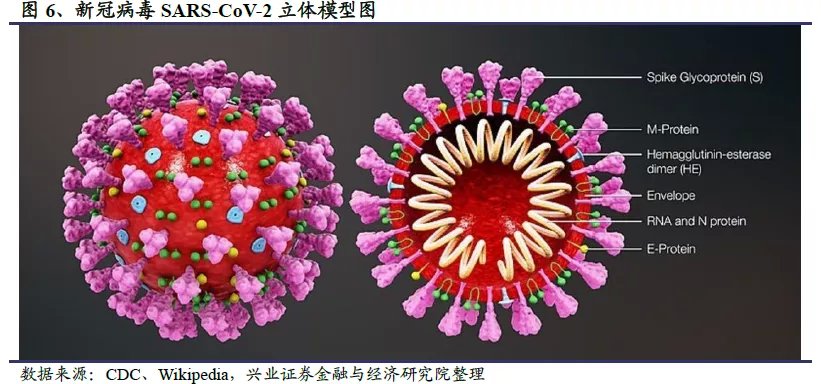
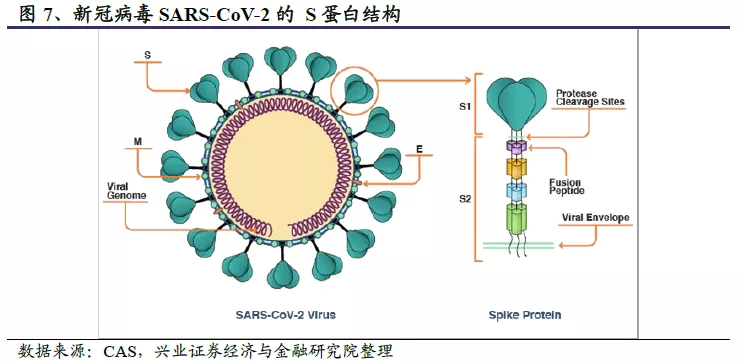
刺突状的S蛋白是公共卫生事件的病毒最重要的表面膜蛋白,决定公共卫生事件的病毒的宿主范围和特异性,是中和抗体及疫苗设计的关键靶点。S蛋白的形状和螺丝钉有几分相似,但头部更大,柄部也更细长。S蛋白以三聚体的形态存在,主要包括三个S1头部亚基和一个三聚体S2亚基。其中S1亚基头部尖端含有受体结合域(Receptor Binding Domain,RBD),可以与人类受体—血管紧张素转化酶2(ACE2)结合。通过RBD的基因重组或突变可以实现不同宿主间的传播,并导致较高的致死率。S2亚基更靠近病毒膜,负责病毒膜和宿主细胞膜的融合。
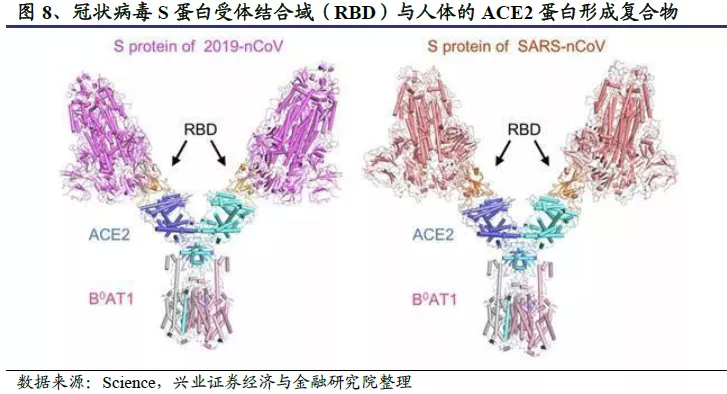
公共卫生事件的病毒入侵细胞时,首先S蛋白RBD与人体细胞的表面受体ACE2结合,随后病毒膜和细胞膜融合,病毒被内化进入细胞,在细胞内进行转录和复制,重新组装成大量新的病毒,又去继续感染其他细胞。有研究表明,不同的SARS-CoV病毒对人血管生成素受体蛋白ACE2的亲和力不同,感染力和传播力也不同。公共卫生事件的病毒RBD与人ACE2的结合能力明显高于SARS病毒与人ACE2的亲和力,原因是公共卫生事件的病毒RBD中发现的F486和N501等关键突变增强了其与人ACE2的结合能力。
2.2、卫生事件现有治疗方案
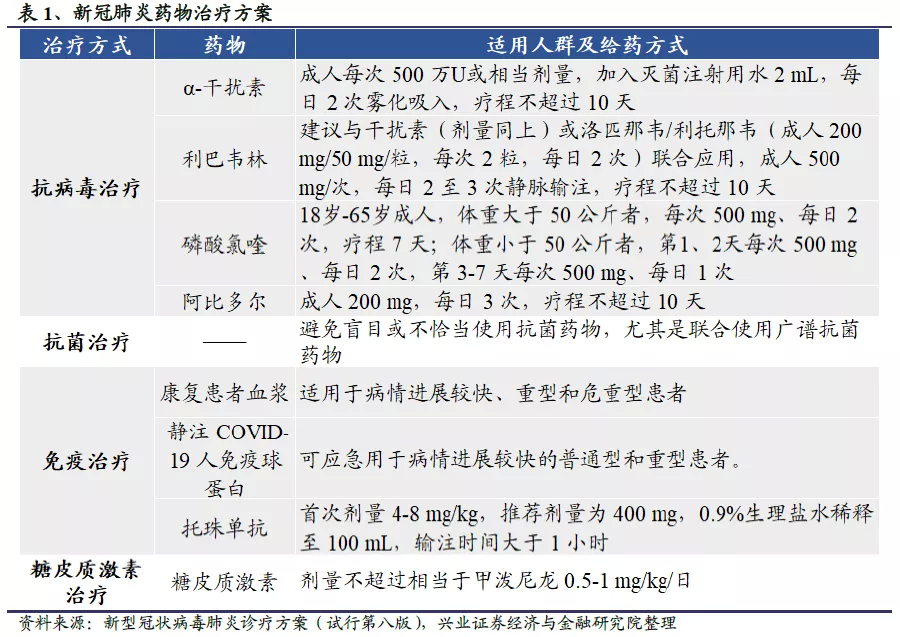
目前卫生事件没有“特效药”,常规治疗手段为“肺炎基础疗法+抗病毒药物+抗炎药物”。根据国家卫健委8月中印发的《公共卫生事件的诊疗方案(试行第八版)》,对于一般患者,多采用供氧治疗与加强支持治疗的方式;对重型、危重型患者则在抗病毒治疗的基础上,针对不同临床表现的患者使用康复患者血浆、免疫球蛋白等免疫治疗或糖皮质激素治疗等其他治疗手段。但总体来说,在没有卫生事件“特效药”的情况下,目前采用的治疗手段大体是“肺炎基础疗法+抗病毒药物+抗炎药物”的思路,其余附加治疗方法依照患者病情进展酌情使用。
如果说现行的卫生事件诊疗方案是基础,那么“预防”及“消灭”卫生事件则要寄希望于正在研发的疫苗和特效药。目前临床在研的卫生事件疫苗及治疗药物主要分为以下4类:
小分子抗病毒药物:化药瑞德西韦、被称为“人民的希望”,其余包括已上市药物洛匹那韦/利托那韦、法匹拉韦等也在进行治疗卫生事件的临床试验。
抗炎药物:多个生物药被用于抑制“炎症因子风暴”,例如托珠单抗(Tocilizumab)、西妥昔单抗(Siltuximab)等;也有部分小分子消炎药的临床试验,例如巴瑞克替尼(Baricitinib)、鲁索替尼(Ruxolitinib)等。
中和抗体:特异性高、副作用小,有望成为治疗卫生事件的“特效药”,其中再生元的“鸡尾酒”疗法REGN-CoV2、礼来×AbCellera的LY-CoV555、VIR×GSK的VIR-7831进展较快,国内企业中君实生物×礼来的JS016领跑。
疫苗:重组蛋白疫苗、核酸疫苗、病毒载体疫苗、灭活疫苗和减毒活疫苗等五条线路并进,国内外核心企业积极布局,部分已进入临床III期。
综合考虑研发时长、研发成功率、药物特异性、安全性等多维度因素,我们认为,中和抗体有望成为治疗卫生事件的“特效药”;疫苗研发成功后,疫苗预防联合抗体药物治疗是终结公共卫生事件的希望。

3、什么是中和抗体?为什么要用中和抗体?
3.1、什么是中和抗体?
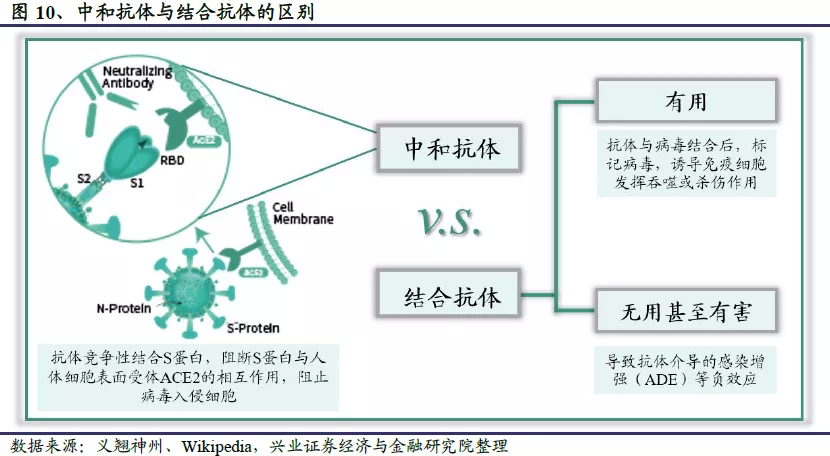
中和抗体是当病原微生物侵入人体时B淋巴细胞产生的一类抗体。当病毒、细菌等病原微生物入侵人体后,会激活人体内的免疫系统,刺激B细胞产生多种抗体。但只有部分抗体能够迅速识别病原微生物,与其表面的抗原结合,阻止该病原微生物结合靶细胞表面的受体侵入细胞,从而保护人体不被感染。这个过程叫做中和作用,发挥作用的抗体称为中和抗体。类似于化学的酸碱中和,中和作用的意思是病毒来了,免疫细胞把中和蛋白分泌到血液里,与病毒颗粒结合,阻止病毒感染细胞,破坏病毒颗粒,把病毒“中和”掉。病原体激活人体免疫系统后,B细胞产生的抗体不仅包括中和抗体,还有结合抗体,结合抗体主要分成有用和无用甚至有害两类。有用的结合抗体能够“标记”病原体,告诉免疫系统这里有“坏人”,诱导免疫细胞将其杀伤和清除;相比之下有些结合抗体可能会发挥反作用,诱导抗体依赖增强(ADE)效应,非但不能抑制反而促进了病毒的感染。因此,中和抗体疗法就是直接用中和抗体进行被动免疫治疗的方法,相当于人为补充“子弹”来支援免疫系统。
3.2、为什么要用中和抗体?
中和抗体是一种大分子药物,与小分子药物相比,具有特异性好,副作用小,近年来已有开发中和抗体药物应对传染性疾病的成功先例。针对埃博拉病毒,两款单克隆抗体药物MAb114和REGN-EB3已经通过临床试验证明能够有效降低患者死亡率,目前美国FDA已授予MAb114突破性药物资格和孤儿药资格,并批准REGN-EB3进行优先审查。还有中东呼吸综合征(MERS),2018年经过基因改造的牛生产的针对MERS病毒的人类抗体,在一期临床试验中表现良好。二期临床因为公共卫生事件消失无法继续。针对艾滋病,2018年,抗体药物Trogarzo获得美国FDA批准上市,与其他抗病毒逆转录的药物联合使用治疗艾滋病。以上这几种抗体药物都是中和抗体。需要注意的是,以PD-1/PD-L1为靶点的抗体药物是近年免疫治疗的研究热点,但PD-1/PD-L1的抗体并不是中和抗体,而是与T细胞相关的抗体,目的是恢复T细胞识别肿瘤细胞的功能,从而增强T细胞杀死肿瘤细胞的作用。
中和抗体具有治疗加预防的双重效果。相比疫苗或抗病毒药物,抗体药物之所以独特,是因为它们既能治疗公共卫生事件的病毒感染,又能预防公共卫生事件的病毒感染。相比“老药新用”的小分子抗病毒药物,中和抗体是针对公共卫生事件的病毒开发的疗法,具有非常高的特异性。同时,抗体在血液中的稳定性较高,注射中和抗体可以提供被动免疫力,达到预防公共卫生事件的病毒感染的效果。在预防性疫苗问世之前,中和抗体可能是为特定高危人群(例如老年人和一线医务工作者)提供免疫力的有效方法。
3.2.1、中和抗体VS疫苗
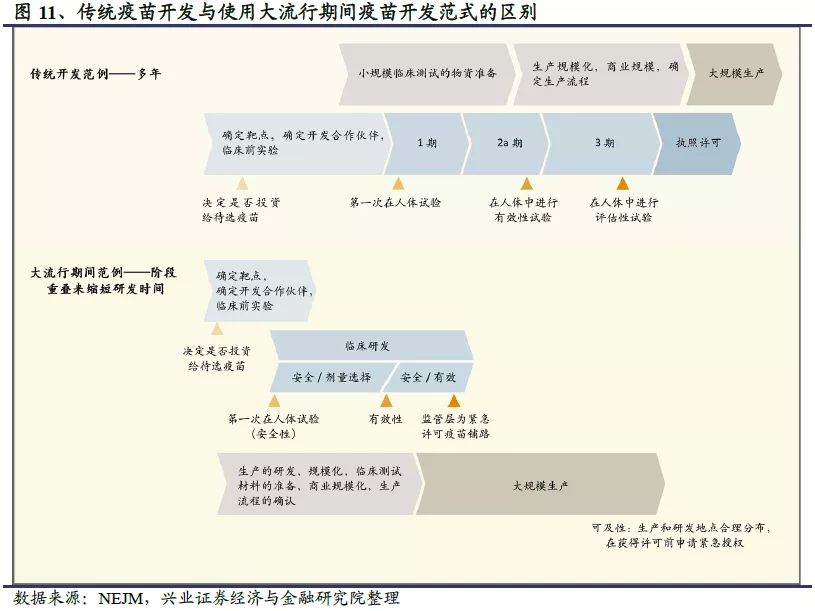
疫苗研发时间较长,短期内无法上市普及。疫苗的开发是一个长期并且昂贵的过程,失败率很高。其中传统疫苗的研发一般遵循线性步骤,普遍需要5-10年的时间;在大流行病期间,因为需要快速开发疫苗可能会平行进行多步骤,但总体而言也需要1-5年的时间。以埃博拉疫苗为例,2014 年埃博拉公共卫生事件于西非大规模爆发,俄罗斯的 GamEvac联合疫苗通过 I/II 期临床研究结果率先获批,时间是2016年,用时2年;随后,中国军事科学院联合康希诺研发的埃博拉疫苗Ad5 -EBOV于2017年10月在国内获批,供应急使用及国家储备;而默沙东的VSV-EBOV疫苗则于 2019 年11 月及 12 月分别获欧盟 EMA 及美国 FDA批准上市,此时距离2014年埃博拉公共卫生事件爆发已经过去5年。对于公共卫生事件而言,进展最快的疫苗研发路线已经进入临床III期试验阶段,其中国产疫苗进展最快的是国药集团旗下武汉生物制品研究所、北京生物制品研究所研发的卫生事件灭活疫苗,有望今年年底上市;美国疾控中心CDC主任表示,卫生事件疫苗在今年底或明年1月上市后将优先供应医务工作者及现场应急人员,预计2021年春夏才能向普通民众发放。因此在疫苗研发广泛普及之前,我们需要强有力的治疗方法、有针对性的特效药,来降低死亡率。
3.2.2、中和抗体VS小分子药物
小分子特效药开发周期长,现下小分子药物多是“老药新用”,并非针对性治疗卫生事件的药物。洛匹那韦/利托那韦、氯喹/羟氯喹等小分子药物虽然被广泛用于抗病毒治疗,但其并不是治疗卫生事件的“特效药”。开发一款针对公共卫生事件的病毒的小分子药物可能需要6至10年甚至更长的时间,所以目前使用的小分子药物多是“旧药”新用。氯喹/羟氯喹原本是用于治疗疟疾的小分子药物,而洛匹那韦/利托那韦是常用的抗艾滋病药物,这也能够解释为何疗效不够理想。被誉为“人民的希望”的瑞德西韦,也并非是专门为公共卫生事件的病毒研发的。2009年,吉利德公司最早针对丙型肝炎研发出瑞德西韦,分子式改进后曾于埃博拉公共卫生事件期间试验过药物安全性及有效性。在试验中,瑞德西韦的安全性得到了验证,但因有效性不及其他两个方案(另外两个方案均为中和抗体),没有被申请注册用于埃博拉病毒的治疗。虽然美国FDA于5月1日批准卫生事件患者在紧急下可以使用瑞德西韦治疗,但其临床表现也没有达到人们心目中理想的“特效药”效果。
3.2.3、中和抗体VS血浆疗法
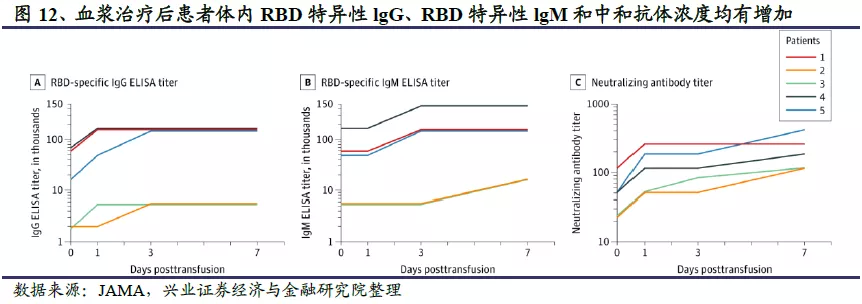
血浆疗法初见成效,但来源有限,不可能大规模使用。除小分子药物外,血浆疗法也初见成效。血浆疗法是把康复者的血浆离心后,取上层血清注射给病人。研究发现,人体感染公共卫生事件的病毒后,一周内会产生IgM抗体,但8-12周后会迅速减少;感染后1-2周会产生IgG抗体,属于中和抗体,在3-4周达到高峰,可持续存在几个月甚至更长时间。因此,血浆疗法的本质是一种抗体疗法,利用卫生事件康复患者血浆中残留的中和抗体治疗其他患者。由于种种限制因素,血浆疗法很难大规模使用。首先,血浆是一个复杂的混合物,有数千万甚至上亿种抗体,里面有具有治疗效果的中和抗体,也有没有疗效甚至其副作用的其他抗体,有效成分的比例比较低;其次,康复者的血浆具有个体差异,输入病人体内有引发排异等副反应的可能性;最后,由于血浆来源有限,主要靠康复者捐赠,很难大规模供应,不能解决全球接近2千万感染者的治疗难题。
中和抗体疗法可以认为是人为筛选和制备了血浆中具有治疗作用的抗体。人类的血浆中大约有数千万至上亿种抗体,对于公共卫生事件的病毒的治疗类似于“万箭齐发”,瞄准公共卫生事件的病毒的同时也可能牵连无辜;而中和抗体是经过人为筛选和制备的抗体药物,成分单一,特异性高,安全性好,可精准靶向公共卫生事件的病毒的抗原位点,是一种良好的治疗和预防手段。
4、中和抗体的开发策略
4.1、中和抗体的临床前研发
据Antibody Therapeutics数据库统计,截至2020年9月,全球共有98个靶向公共卫生事件的病毒S蛋白的中和抗体项目。在此我们以研发进度较快、关注度较高的部分中和抗体为例,分析中和抗体在临床前研发阶段、临床试验阶段及生产阶段应关注的主要问题。
4.1.1、中和抗体的靶点选择
病毒S1亚基的RBD与人类受体ACE2的结合是介导病毒感染宿主细胞的重要环节,因此目前公共卫生事件的病毒中和抗体药物研发的靶点大多集中在S1亚基,尤其是S1亚基的受体结合域RBD。S1亚基可分为两个相对独立的区域,除了含有RBD的C末端结构域(CTD)外,还有一个N末端结构域(S1-NTD),也有少数团队在开发靶向S1-NTD的中和抗体,例如军科院陈薇院士团队和西湖大学周强团队合作开发的中和抗体4A8。
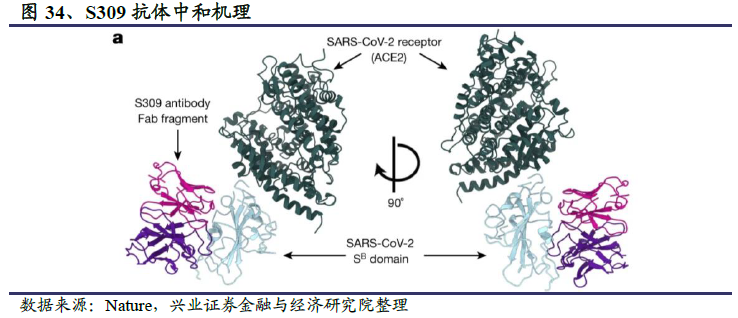
靶向人类冠状病毒的广谱中和抗体不仅能够帮助我们对付当前的公共卫生事件,对于预防和治疗未来未知的冠状病毒也具有重大意义。目前已经报道的广谱中和抗体基本都是靶向S1亚基RBD,典型代表是和铂医药与荷兰乌得勒支大学共同发现的47D11,以及Vir Biotechnology与华盛顿大学筛选出单克隆抗体S309。相比于S1亚基的高变异性,S2亚基更加保守,可能是未来筛选广谱中和抗体时需要关注的潜在靶点。
公共卫生事件的病毒S蛋白表面有近66个糖基化修饰位点,在这些密布的糖基簇拥下,局部有少许蛋白裸露出来,也就是多数在研中和抗体的蛋白靶点。除了蛋白靶点之外,聚糖靶点也值得深入研究。VIR分离得到的S309抗体能够识别RBD上含有糖基化的表位,说明S蛋白的糖基化也是公共卫生事件的病毒中和抗体研发中需要关注的。
4.1.2、中和抗体的作用机理
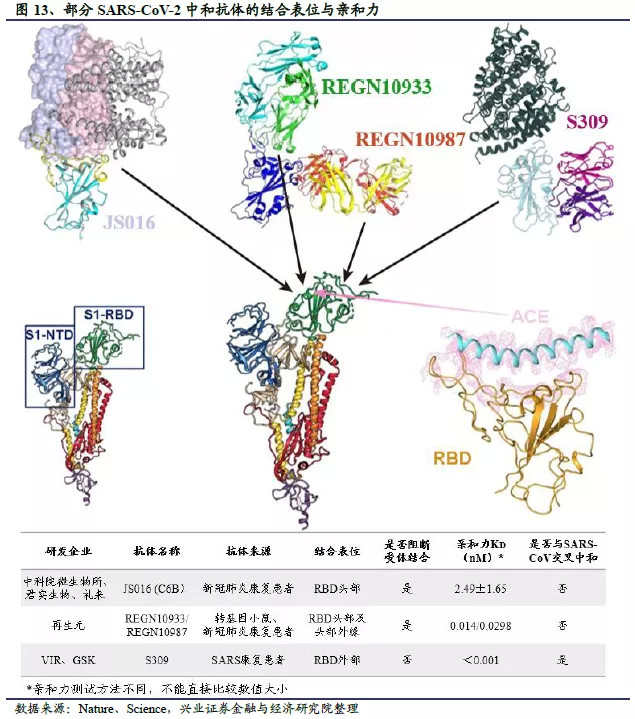
公共卫生事件的病毒中和抗体的最常见的作用机制是靶向病毒S1亚基的受体结构域RBD,阻断RBD与宿主细胞表面ACE2受体结合。考虑到中和抗体要与宿主细胞表面ACE2受体竞争性结合体内游离病毒,因此在亲和力(Affinity)上的基本要求就是要比ACE2更强的结合在病毒S蛋白RBD结构域上,以达到排他的阻隔效果,使病毒无法与宿主细胞受体ACE2结合,防止病毒感染细胞。通常使用平衡解离常数KD表征结合亲和力,KD越小,表明病毒S蛋白和中和抗体结合达到平衡后解离的越少,亲和力越强。体外测得ACE2与公共卫生事件的病毒S蛋白的亲和力常数(KD)在10 nM左右,所以我们保守估计有效的中和抗体亲和力常数应该在1 nM。不过需要注意的是,此处不同研究团队采用的是不同的靶标和不同检测方法得到的结果,不应简单进行绝对数值大小的比较。
此外,Vir Biotechnology和华盛顿大学研究团队的S309,以及和铂医药与荷兰乌得勒支大学共的47D11是目前发现的少见的具有SARS病毒和公共卫生事件的病毒交叉中和性的广谱中和抗体。它们的共同特点是并不直接作用于宿主细胞表面ACE2受体的结合位点,即并不阻断S蛋白与ACE2受体的结合。荷兰乌得勒支大学发表在Nature Communication 上的研究表明47D11对SARS病毒和公共卫生事件的病毒S蛋白的RBD均有结合活性,但没有同ACE2的竞争活性。Vir Biotechnology和华盛顿大学关于筛选出的中和抗体S309的鉴定和表征结果发表在Nature上,研究人员通过冷冻电镜解析蛋白三维结构表明S309的结合位点为RBD结构域外侧的保守区域,与ACE2的结合位点不重合,SPR法也未观察到ACE2的竞争活性。该研究同时报道了S309介导的细胞毒作用(ADCC)和细胞吞噬作用(ADCP),表明S309可能存在多种抗病毒机制。总体而言,这类广谱中和抗体并不阻断病毒S蛋白与人类ACE2受体的结合,而是通过其他机理,比如通过别构效应影响S蛋白对ACE2受体的识别,或影响病毒S蛋白与ACE2受体结合后的构象变化,或阻止病毒膜与细胞膜的融合等方式阻止病毒感染细胞。随着研究的深入,广谱中和抗体的作用机制和临床有效性将得到进一步验证。
4.1.3、中和抗体的筛选平台
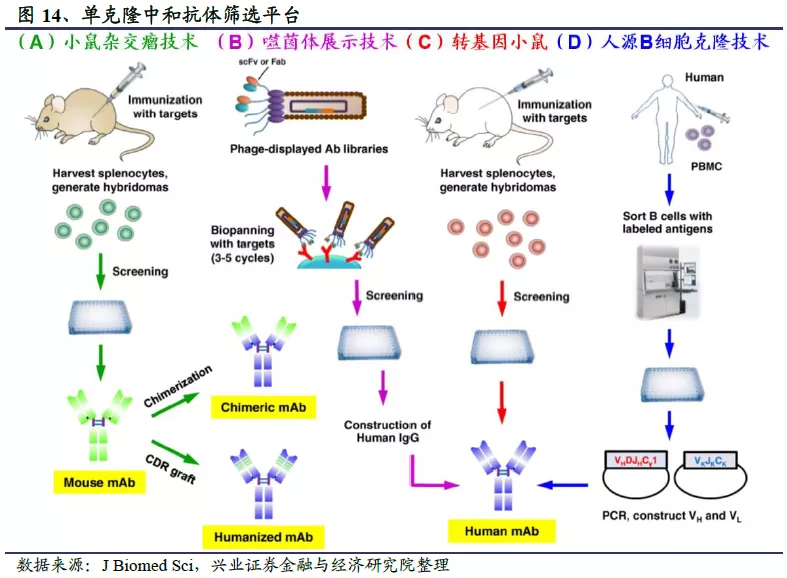
目前中和抗体的筛选主要通过三种技术手段。一是从康复病人的血液里用单B细胞克隆技术筛选中和抗体,先对康复患者的B细胞进行分选,获得产生中和抗体的单个B淋巴细胞,然后通过分子生物学方法获得编码抗体lgG的基因序列,在体外重组表达出中和抗体。这是目前获得单克隆中和抗体最快速有效的技术,目前已经进入临床阶段的中和抗体项目中,绝大多数单克隆中和抗体都是通过这种方式获得的,例如Vir Biotechnology的S309、君实生物的JS016、礼来的LY-CoV555、Tychan的TY027以及腾盛博药的两个中和抗体BRII-196/198等。通过这种方式筛选的中和抗体都有自己各自的特点,由于各研究团队接触的患者各不相同,再加上患者间具有基因多样性,两个团队筛选出相同抗体的可能性非常小。在实际操作中,从康复患者体内提取B细胞的时间点是一个重要因素。相对晚一些的采样时间,能够给予中和抗体足够的亲和成熟时间,Vanderbilt大学团队的研究表明,患者出现症状后的第50天或许是最佳的样本采集时间。其它因素比如患者症状的持续时间和严重程度也会影响采集到的中和抗体的中和能力。
二是通过向动物模型注射病毒S蛋白,激发中和抗体的产生,这类技术的典型代表为再生元制药及和铂医药。再生元基于VelocImmune转基因小鼠平台和卫生事件康复患者血液这两条途径初步筛选出数百个全人源中和抗体,最终选择REGN10933和REGN10987组成抗体鸡尾酒REGN-COV2,但再生元并没有指出这两者哪一个来自小鼠平台、哪一个来自康复患者。和铂医药等开发广谱中和抗体47D11利用的是H2L2全人源转基因小鼠技术平台,体外实验证实47D11对SARS-CoV和SARS-CoV-2都具有中和活性。
三是对曾受到公共卫生事件的病毒感染的康复者血清中的抗体进行筛选,发现具有强力中和作用的抗体。已经进入临床阶段的中和抗体项目中,韩国Celltrion公司的中和抗体CT-P59即来源于康复患者血清抗体库的筛选:Celltrion首先获得了康复患者的血清样本,然后从患者血清中提取DNA,通过DNA转染培养初步鉴定并建立起含有300个候选抗体的样本库,再从中选择最有效的候选抗体开展动物实验和临床试验。
4.1.4、如何防止因病毒突变导致的中和抗体失效
防止因病毒突变导致中和抗体药物失效是临床前开发的难点。中和抗体药物大多靶向S蛋白的RBD区域,如果SARS-CoV-2病毒在快速复制传播的过程中靶点发生了突变,抗体药物可能面临失效,为此各公司研发过程中都采取了不同的方式降低中和抗体失效的可能性。其中一种策略是采用多种抗体的组合,例如再生元使用鸡尾酒疗法,结合两种抗体REGN10933和REGN10987,这两款抗体结合RBD的不同位点,因此组合使用可以降低病毒通过基因突变,逃避单抗体治疗的可能性。阿斯利康的AZD7442也是由2种单克隆抗体AZD8895 和 AZD1061组成的鸡尾酒疗法。礼来在对LY-CoV555开展治疗轻至中度患者的II期临床试验中,也引入了从君实生物获得授权的LY-CoV016进行联合给药,探索单抗鸡尾酒疗法的治疗效果。另一种策略是选择相对保守的抗原位点,例如Vir Biotechnology的S309靶向的是SARS-CoV-2病毒上高度保守的表位,这种类型的抗原表位通常位于病毒抗原结构的关键部位,产生突变可能会影响到抗原的正常功能,削弱病毒的传播能力,因此其通过产生突变逃避抗体治疗的可能性相对较小。
4.1.5、抗体介导的感染增强效应(ADE)
抗体依赖增强(antibody dependent enhancement,ADE)是指非中和抗体或低亲和力的中和抗体促进病毒感染,引起感染增强的作用,这种现象在登革病毒、SARS-CoV病毒等多种病毒都均有报道。ADE的相关机制比较复杂,其中一个作用机制是中和抗体的Fc片段与巨噬细胞表面的FcγR受体结合导致抗体被巨噬细胞内吞,这样抗体反而为病毒入侵细胞提供了的一个新途径,加重了病毒的感染。另一个作用机制的结构依赖的感染增强,当抗体与S蛋白结合后,使其处于一种更易于发生膜融合的状态,进而增强病毒的感染。
我们认为目前还不需要太过担心公共卫生事件的病毒中和抗体的ADE效应。一方面暂时没有明确关于在研中和抗体ADE效应的明确报道,科学家们只是根据SARS-CoV-2与SARS-CoV的S蛋白有较高的同源性进行的理论推测。另一方面中和抗体的给药浓度相对较高,通常大于10 mg/kg(埃博拉抗体给药剂量为50mg/kg)。ADE一般在非中和抗体或低亲和力的中和抗体才会产生,高亲和力的中和抗体在高浓度下产生ADE效应的可能性极低。
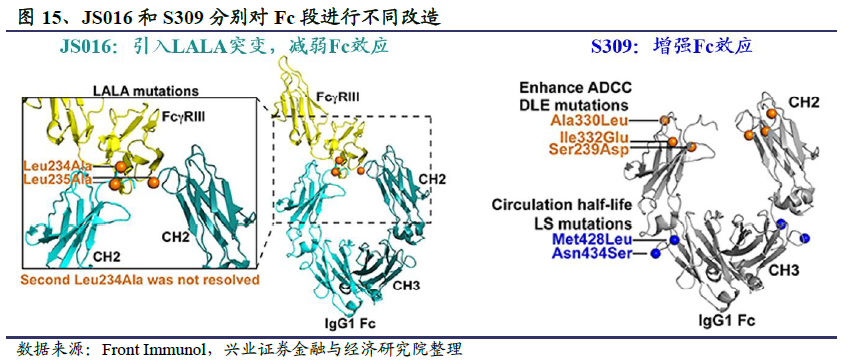
此外,假如卫生事件中和抗体确实能够引发ADE效应,有研究团队已经考虑到了这种风险并通过抗体Fc片段改造减少这种负面影响。君实生物和中科院微生物所对筛选出的中和抗体CB6(JS016)的Fc段进行了工程改造,引入LALA突变以避免可能出现的抗体介导的ADE效应,同时还能降低抗体介导的细胞毒性作用(ADCC),降低急性肺损伤风险。但有意思的是,ADCC在清除病毒感染的宿主细胞时起重要作用。考虑到SARS-CoV-2在宿主细胞内会产生大量的病毒包涵体,引入抗体的ADCC作用可以快速清除病毒感染的宿主细胞,从而能够加速患者体内的病毒清除,因此Vir Biotechnology选择了截然相反的方向,对中和抗体S309进行了Fc段功能加强,旨在通过ADCC和ADCP作用加强抗病毒效果。总之,如何对待ADE问题,以及究竟应该选择哪个方向,应该从抗体本身的功能和应用出发,具体问题具体对待。
4.2、中和抗体的临床实验
公共卫生事件的病毒单克隆中和抗体的临床试验同样分三期进行:I期临床试验的主要目的是评价候选抗体的安全性、耐受性和药代动力学等;II期临床试验会对治疗效果做初步评估;III期临床试验会针对多个适应症进行,包括高风险感染者的预防、轻至中度患者治疗和重症住院患者治疗等。
I期临床试验:初步的临床药理学及人体安全性评价试验。观察人体对于新药的耐受程度和药代动力学,为制定给药方案提供依据。公共卫生事件的病毒中和抗体的I期临床试验最快不少于20天,入组人数较少,一般为10-50例。预期公共卫生事件的病毒中和抗体有较好的安全性和耐受性,治疗窗口较宽。
II期临床试验:治疗作用初步评价阶段。目的是初步评价药物对目标适应症患者(可能根据病情轻重、年龄、种族等进行分组)的治疗作用和安全性,也包括为III期临床试验研究设计和给药剂量方案的确定提供依据。此阶段的研究设计可以根据具体的研究目的,采用多种形式,包括随机盲法对照临床试验。II期临床试验最短需要1个月的时间,入组病例数比I期多,一般为100-500例。
III期临床试验:治疗作用确证阶段。目的是进一步验证药物对目标适应症患者的治疗作用和安全性,评价利益与风险关系,最终为药物注册申请获得批准提供充分的依据。试验一般应为具有足够样本量的随机盲法对照试验。III期临床试验最短需要3-6个月的时间,入组病例数更多,一般为1000-3000例。
公共卫生事件的病毒中和抗体的III期临床试验针对多个适应症进行布局,主要分为治疗试验和预防试验。
治疗实验:根据患者病情分为轻症治疗和重症治疗,轻症治疗临床试验入组的受试者为轻至中度、非住院治疗的患者;重症治疗临床试验入组的受试者为重症住院的患者。一个疗程(约4周)结束后即可揭盲。
预防试验:目的是验证中和抗体预防高风险感染人群中感染卫生事件的有效性和安全性,分为暴露前预防和暴露后预防。暴露前预防一般8周内可获得初步试验结果,暴露后预防1个月内可获得初步结果,研究完成需要随访6-8个月。
4.3、中和抗体的研发与生产
SARS-CoV-2中和抗体药物研发最重要的是速度快。在公共卫生事件结束之前上市的中和抗体药物,才最具有价值,因此中和抗体的研发是一场争分夺秒的赛跑,不仅要抢在病毒前面,也要在有限但激烈的研发竞争中走在前列。公共卫生事件的病毒中和抗体面临的是一个“短期存在的蓝海市场”,存在“赢家通吃”的特征,只有跑的最快的少数几家才有可能最先完成临床试验并获批上市。
中和抗体研发的关键在于临床试验的速度和效率。中和抗体的临床前筛选过程更偏工程,科学上的挑战难度不大,因此各研发团队用时均在1-2个月之间,没有造成明显的时间差。在这种情况下,如果想在激烈的研发竞争中走在前列,开展临床试验的时间和进行临床试验的效率就显得尤为重要,谁能先进入临床试验就有机会抢占先机,谁能先结束临床试验就有机会优先上市,独享百亿美元级蓝海市场。因此各个团队比拼的是开展临床试验的资源,这方面再生元、礼来等成立时间久、临床试验经验丰富的企业将具有明显优势。随着国内公共卫生事件的有效控制,君实生物后续预防和治疗临床试验将需要在海外开展。
中和抗体的规模化生产能力也至关重要。随着疫苗研发的快速推进,我们预计中和抗体用于预防的时间窗口为上市后到2020年底;而疫苗上市后,随着群体免疫的逐步形成,公共卫生事件或得到控制,我们预期中和抗体用于治疗的时间窗口为上市后到2021年底(假设公共卫生事件持续到2021年底或更久)。因此除了关注中和抗体的获批时间,获批后能否在短时间内大规模供应也显得非常重要。本身具有抗体药物规模化生产工艺的企业将具有一定优势,例如君实生物、再生元、礼来等。礼来已做好年底生产10万剂LY-CoV555的准备,且9月16日宣布将与安进合作生产中和抗体药物以增强供应能力。再生元7-30万治疗剂量(或42-130万预防剂量)的REGN-CoV2预计秋季生产完成,且8月20日再生元已宣布与罗氏达成抗体药物的生产合作,生产能力将至少提升3.5倍。另外也有企业通过和CDMO公司合作完成大规模生产,例如Vir Biotechnology已经与Biogen、三星生物签订合约,临床试验结果理想的情况下,预计20201年生产1000 -1500万剂中和抗体药物。
5、中和抗体的市场空间
5.1、公共卫生事件持续发酵,确诊人数持续飙升
截至2020年9月19日,全球现有卫生事件确诊人数737万人,进入9月以来平均每天新增确认28万人。全球公共卫生事件一波未平一波又起,从1月底最先在国内爆发,至3月初肆虐欧洲,之后美国、巴西、俄罗斯、南非、印度等国家的公共卫生事件都曾经或正在迅速蔓延。此外,部分公共卫生事件控制较好的国家,例如西班牙、法国、英国等欧洲多国,随着复工复产的开始,反弹迹象明显。WHO表示,随着全球卫生事件确诊人数与死亡病例持续飙升,全球距离公共卫生事件结束仍十分遥远。
欧美发达国家中,美国每日新增确诊人数3-5万人;欧洲、加拿大、日本等其他发达国家每日新增确诊人数约3万人,其中欧洲公共卫生事件反弹迹象明显,未来确诊人数还会进一步增加。整个欧美发达国家市场每日新增确诊人数6-8万人。
巴西、印度等发展中国家中,印度每日新增确诊人数9-10万人;巴西每日新增确诊人数3-5万人。整个发展中国家国家市场每日新增确诊人数约20万人。
5.2、中和抗体兼具预防与治疗作用
中和抗体兼具预防与治疗的双重作用。中和抗体的预防作用主要是保护两类高危人群——超过五六十岁的老年人和每天接触患者的医护人员;同时需要注意的是,中和抗体只能保证短期预防,需要每1个月或者每2个月打一针,要想做到全民免疫还是要等到疫苗研发出来。发挥治疗作用时,参考埃博拉抗体药物REGN-EB3和MAb114在降低死亡率方面均比瑞德西韦要好,卫生事件中和抗体药物的治疗效果也应该会好于瑞德西韦。此外,中和抗体安全性好,给药方式也更加方便,只需要一次性皮下注射或静脉注射,无需重复给药。
5.3、从需求端与成本端看中和抗体定价
7月美国政府花费4.5亿美元从再生元购买了相当于7 -30万治疗剂量,或42 -130万预防剂量的REGN-COV2鸡尾酒抗体药物,据此订单价格估算,卫生事件用于患者治疗时单剂价格为1500-6430美元,用于高风险者预防时单剂价格为350-1070美元。另一方面,参考瑞德西韦在供给美国政府的定价为390美元/瓶,针对美国私立保险公司的价格为520美元/瓶,预计绝大多数患者将接受5天疗程的治疗,使用6瓶瑞德西韦,即每位患者一个疗程需要花费2340-3120美元。成本方面,君实生物COO表示,中和抗体用于预防时每剂的蛋白量达到1-3克,单抗规模化生产成本约为100美元/克左右,则每剂中和抗体的生产成本为100-300美元;根据行规,抗体药售价是其成本的5倍左右,则从成本端估算,中和抗体用于预防时单剂价格为500-1500美元,价格范围与美国政府给予再生元的订单价格非常接近。以上估算主要基于美国等发达国家,在巴西、印度等公共卫生事件严重的发展中国家,卫生事件中和抗体的价格要远低于欧美发达国家,例如PD-1抑制剂帕博利珠单抗在国内售价仅为美国的54%。吉利德公司也表示在发展中国家将以“大大降低的成本价”提供瑞德西韦,目前已有两款瑞德西韦仿制药在印度批准上市,分别定价39-52美元/瓶和65-78美元/瓶,仅为原研药价格的10%-20%,由此推测中和抗体药物在发展中国家定价有望降至发达国家的20%-40%。
因此我们假设,在欧美发达国家市场,卫生事件中和抗体的用于治疗时单剂价格为1000-2000美元,用于预防时单剂价格为350-800美元;在印度、巴西等发展中国家市场,卫生事件中和抗体用于治疗的单剂价格为400-1000美元,因中和抗体价格相比疫苗较为高昂,暂不考虑发展中国家的预防市场需求;公共卫生事件特殊时期、特殊地区,定价可能根据实际情况而下调。
5.4、百亿美元级市场,空间广阔
核心假设:
欧美发达国家每日新增确诊人数6-8万人,假设每日新增治疗需求2万人。预防性中和抗体主要针对高风险人群,假设高风险人群与新增确诊人数比为1.5:1(密切接触的家人、老年人及医护人员等),即每日新增预防需求3万人。进度最快的再生元有望在秋季完成美国政府首批订单,因此我们假设欧美国家对中和抗体药物的需求从11月开始逐步被满足。
印度、巴西等发展中国家每日新增确诊20万人,因为今年底之前中和抗体将在发达国家市场上供不应求,所以只考虑公共卫生事件持续到2021年时发展中国家的治疗需求。
预计疫苗的获批时间会晚于中和抗体,国产疫苗中进度最快的灭活疫苗有望今年年底上市;美国疾控中心CDC主任表示,疫苗有望今年底或明年1月在美国上市。但疫苗上市后将优先供应医务工作者及现场应急人员,普通群众22021年春夏才能广泛接种疫苗。考虑到疫苗接种率的提高需要一定时间,我们假设公共卫生事件将会持续到2021年末。由于疫苗能够提供主动免疫,预防效果更好,而且价格会远低于中和抗体,因此我们预计疫苗上市后发达国家的预防市场会被疫苗替代,治疗需求方面,随着疫苗在健康人群中的覆盖率逐渐提高,卫生事件新增确诊人数将会逐渐下降,因而我们假设疫苗上市后发达国家治疗需求减半,即每日新增治疗需求1万人,目前发展中国家每日新增确诊是发达国家的2-3倍,但考虑到抗体药价格较高,假设发展中国家每日新增治疗需求同样为1万人。
中和抗体的放量速度方面,现有的研发进度较快且披露生产计划的企业包括再生元、礼来和VIR。再生元7月初获得美国政府7-30万剂治疗剂量(或42-130万预防剂量)的订单,预计秋季生产完成;8月与罗氏合作后,再生元生产抗体药物的能力将至少提升3.5倍。礼来方面,临床试验结果理想的情况下,也预计年底前至少上市10万剂LY-CoV555。VIR预计2021H1开始提供VIR-7831/7832,总量将达到1000-1500万剂。我们认为,2020年底前,再生元和礼来等走在前列的企业将有望提供50-100万剂治疗剂量(或100-200万预防剂量)的中和抗体药物。
公共卫生事件持续到2020年末的情况下,我们预计中和抗体的商业化市场空间约18.3-38.4亿美元,实际市场空间主要取决于中和抗体的定价及各企业的放量速度;假设公共卫生事件持续到2021年末,我们预计中和抗体的市场空间可达68.7-146.4亿美元。其中欧美发达国家市场空间更大,达到54.3-110.4亿美元;发展中国家市场规模约14.4-36.0亿美元。另一方面,中和抗体用于患者治疗的市场空间更大,约62.4-132.0亿美元;用于高风险人群预防时,因为价格高、预防时间短等劣势,疫苗研发成功后很容易被替代,市场规模相对较小,或将不超过10亿美元。由此可见,中和抗体的市场空间非常广阔,而且赛道玩家比较少,全球仅有礼来、再生元、Vir Biotechnology、君实生物等少数几家公司走在前列。
6、海内外研发卫生事件中和抗体的重点公司
据Antibody Therapeutics数据库统计,截至2020年9月,全球共有98个靶向公共卫生事件的病毒S蛋白的中和抗体项目,其中12个候选中和抗体药物进入临床研究阶段。
全球范围内,进展速度较快的中和抗体项目包括由再生元(Regeneron)公司开发的抗体鸡尾酒REGN-COV2,礼来和加拿大AbCellera公司联合开发的中和抗体LY-CoV555,均已进入预防和治疗临床试验阶段。Vir Biotechnology联合葛兰素史克开发的两种候选单克隆中和抗体VIR-7831和VIR-7832,VIR-7831已经于8月底进入临床II/III期试验,VIR-7832也预计将于下半年进入临床试验。
国内公司中,君实生物和中科院微生物所联合开发的JS016、腾盛博药的两款中和抗体BRII-196和BRIII-198、迈威生物的MW33和神州细胞的SCTA01已进入临床试验阶段。其中君实生物的JS016领跑,是中国第一个、全球第二个进入临床试验的公共卫生事件的病毒中和抗体疗法。该候选药物也受到的礼来公司的青睐,双方于今年5月4日达成协议,礼来以最高2.55亿美元的价格买断JS016大中华区以外的临床开发和商业化权益。
6.1、再生元:中和抗体鸡尾酒疗法鼻祖,已有埃博拉抗体成功先例
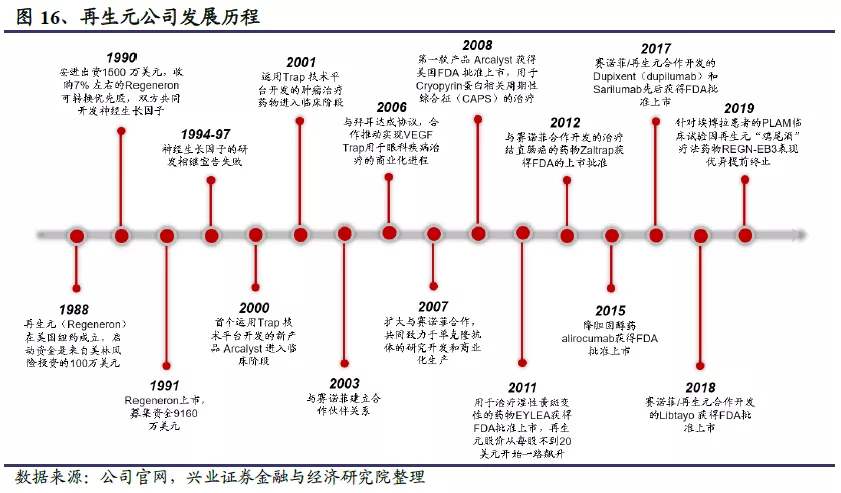
1988年,再生元(Regeneron)公司由伦纳德•施莱弗尔和乔治•雅克波罗斯创立于美国纽约;1991年,公司在纳斯达克挂牌上市。成立之初,再生元的定位是研究神经生长因子,试图开发治疗神经系统疾病的药物,但先后均已失败告终。之后公司调整战略方向,开发出“Trap”技术平台,并基于此贡献了著名的眼科药物EYLEA。同时,再生元还开发并整合了另一个值得称道的技术平台VelociSuite®,用于发现、开发和生产人源单克隆抗体,并以此为基础研发出Praluent,Dupixent,Kevzara和Libtayo等成功药品。发展至今,再生元已经成为一家从事药物发现、开发、生产和销售的集成化生物制药公司,拥有员工8100人,拥有7款美国FDA批准上市的药品。
6.1.1、技术平台
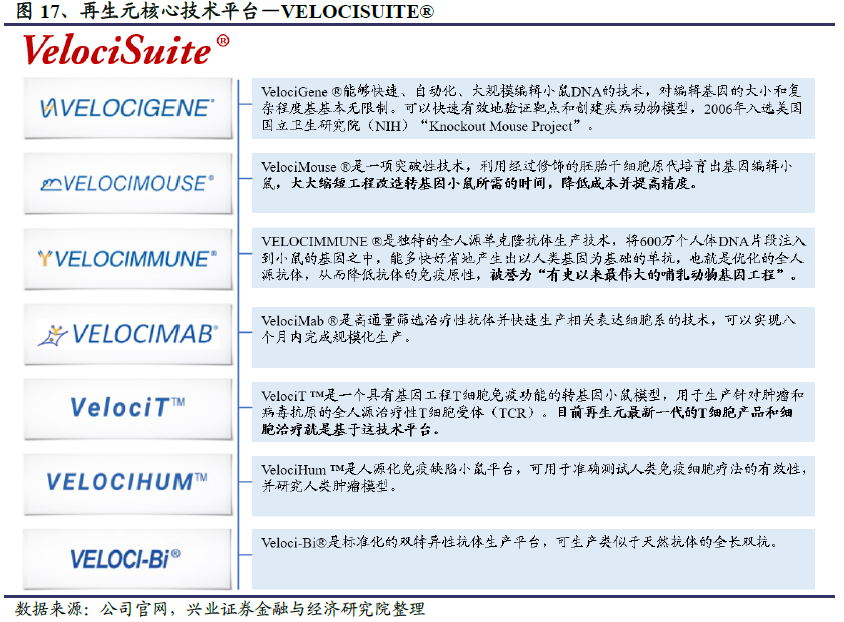
再生元主要有三大核心技术平台,分别是Trap技术平台、VelociSuite®技术平台和Cell Production技术平台。再生元卫生事件抗体研发项目主要利用VelociSuite®技术平台中的VelocImmune的小鼠模型。
VelociSuite®技术平台用于发现、开发和生产人的单克隆抗体,并提高单克隆抗体的生产效率,核心优势是能够简化抗体药物的研发工艺并提高抗体新药的研发品质。VelociSuite®技术平台由VelociGene、VelociMouse、VelocImmune、VelociMab、VelociT、VelociHum和Veloci-Bi七部分组成。VelocImmune是其中最为核心的技术,用于生产全人源单克隆抗体,作用原理是通过将人类基因片段注入到小鼠体内,多快好省地产生出以人类基因为基础的单抗,也就是优化的全人源抗体,从而降低抗体的免疫原性,被誉为“有史以来最伟大的哺乳动物基因工程”。此外,VelociGene能够快速、自动化编辑产生目标基因或高度定制化的基因,VelociMouse用于培育原代胚胎干细胞小鼠,VelociMab用于快速筛选抗体和快速生产表达细胞系。
6.1.2、中和抗体项目
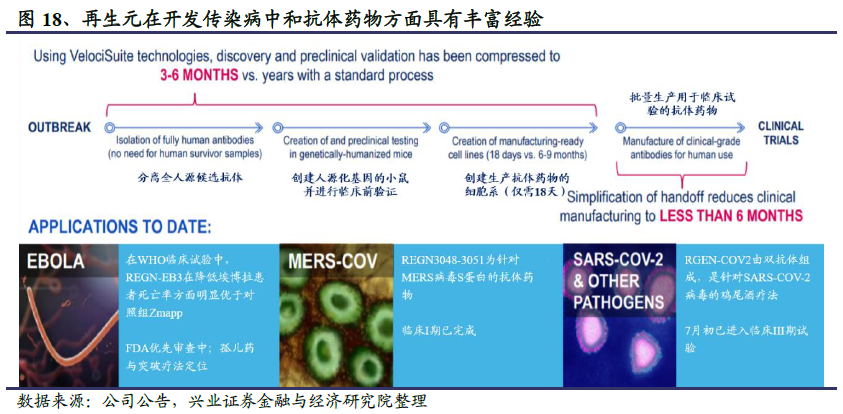
再生元拥有全球领先的全人源抗体转基因小鼠技术平台,最快可在6个月内完成临床前研究、抗体筛选以及生产。目前再生元已用该技术成功研发了埃博拉病毒中和抗体药物REGN-EB3、MERS病毒中和抗体RGEN3048-3051,因此其针对卫生事件的鸡尾酒抗体疗法也备受期待。
1)埃博拉抗体药物REGN-EB3
1976年至2018年间,世卫组织WHO共报告了20多起埃博拉公共卫生事件,其中2014年爆发的埃博拉公共卫生事件最为猛烈,约有29000人感染,超过11300人死亡。为彻底攻克这一恶性传染病,全球众多医疗机构付出了巨大努力,最终产生了四款候选药物:
第一款名为MAb114,是由美国国家过敏症与传染病研究所(NIAID)开发的一种单克隆抗体,是从1995年刚果埃博拉公共卫生事件中的一个幸存者血液中分离而来的。2018年12月,位于迈阿密的生物科技公司Ridgeback Biotherapeutics宣布,从美国NIAID手中买下了MAb114单抗的权利。MAb114已被美国FDA授予孤儿药和突破性疗法,其作用机制是通过和埃博拉病毒的糖蛋白(Glycoprotein,GP)相结合,从而阻断埃博拉病毒和人体细胞表面受体的结合。
第二款名为ZMapp,由3种嵌合体单抗组成,其中两个抗体由加拿大国家微生物实验室开发,另一抗体则来自于美国陆军传染病研究所。该疗法后来被授权给Mapp公司。
第三个名为REGN-EB3,是再生元公司利用其抗体研发平台在六个月内就完成开发的创新疗法。它是一款由3种单抗(REGN3470、3471和3479)组成的鸡尾酒疗法。采用3个抗体是因为埃博拉病毒在不同爆发中会发生突变,这三个抗体每种都能结合到病毒的不同位点,从而从整体上增强鸡尾酒疗法对付突变的埃博拉病毒的能力。
第四个名为瑞德西韦(Remdesivir),即在卫生事件期间人尽皆知的“人民的希望”,是吉利德公司开发的小分子抗病毒药物。它是一种前药,可代谢成为活性的GS-441524,能够起到干扰病毒中的“RNA依赖RNA聚合酶”(RNA-dependent RNA polymerase)的作用。
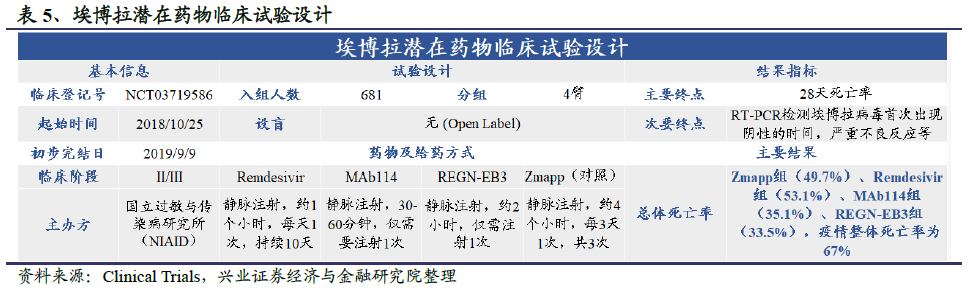
2018年末,WHO启动了一项名为“PALM”的大型随机临床试验,入组681名患者。2019年11月,对于499名患者的初步试验结果发表在新英格兰医学杂志上,结果表明:在接受治疗的第28天,实验组总体死亡率分别为REGN-EB3(33.5%),MAb114(35.1%),ZMapp(49.7%)和Remdesivir(53.1%);对于低病毒载量的患者,死亡率分别是REGN-EB3组(11.2%),MAb114(9.9%),ZMapp(24.5%)和Remdesivir(29.0%)。目前,整个埃博拉公共卫生事件的死亡率为67%。基于此研究结果,可以看出REGN-EB3和MAb114优于另外两款药物,将进一步进行后续的评价。
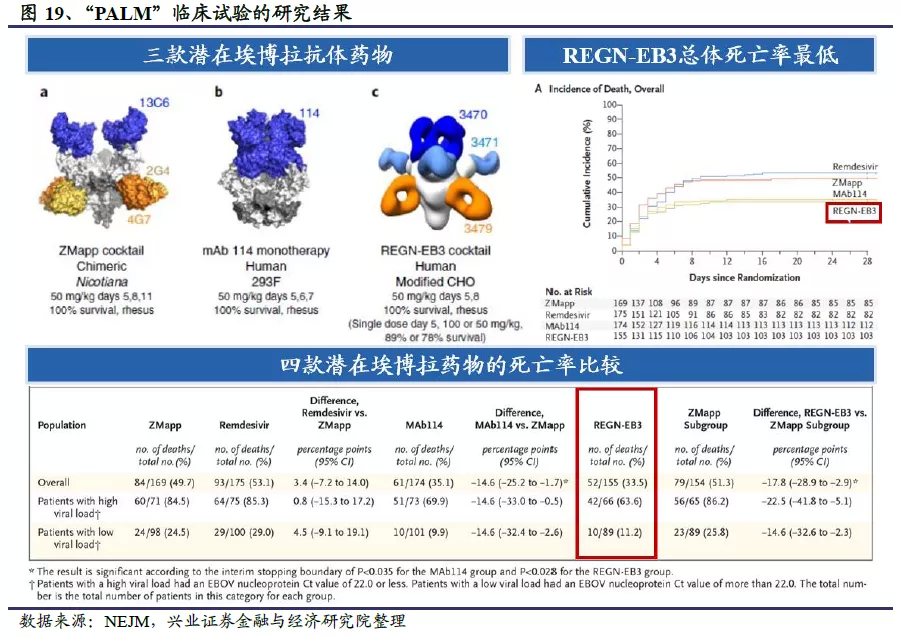
2)公共卫生事件的病毒鸡尾酒抗体REGN-COV2
公共卫生事件爆发后,2020年2月3日,再生元宣布与美国卫生和公共服务部(HHS)合作,共同研发针对公共卫生事件的病毒SARS-CoV-2的治疗性抗体药物。根据再生元公司披露的预期进度,3月已经完成预防和治疗性中和抗体的筛选,4月份推进动物实验,6月开始小规模生产,进行初期临床试验;并在8月份扩大生产规模,达到每月几十万份的预防剂量或几万份的治疗剂量。
再生元的公共卫生事件的病毒抗体药物名为REGN-COV2,是由REGN10987和REGN10933两个单抗组成的鸡尾酒抗体药物。再生元基于VelocImmune转基因小鼠模型和卫生事件康复患者血液这两条途径初步筛选出200多个候选抗体,最终根据候选抗体和S蛋白的结合能力、中和能力以及三维结构表征等参数筛选出两个最合适的单抗组成“鸡尾酒”疗法。REGN10987和REGN10933非竞争性且同时与SARS-CoV-2病毒S蛋白的关键受体结合域(RBD)结合,阻断病毒RBD与人体细胞受体ACE2的结合界面。其中REGN10933靶向处于ACE2结合界面边缘的刺突样环状区域,REGN10933从上方结合病毒RBD,极大的阻碍了病毒RBD和宿主细胞ACE2的结合;而REGN10987从正面和左下方结合病毒RBD。
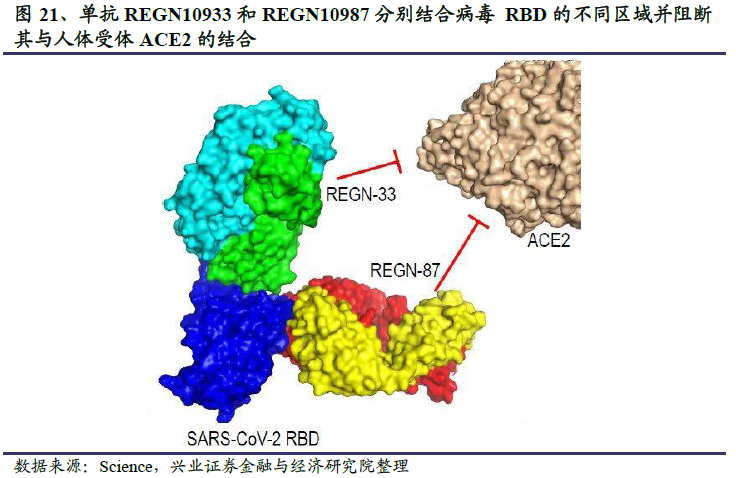
当使用单个抗体进行治疗时,病毒可能会产生多种基因突变逃避抗体的阻断作用,对于结合高度保守区域的抗体也是如此。另一项发表在Science上的研究表明,使用REGN-COV2鸡尾酒抗体治疗公共卫生事件的病毒时,由于REGN-COV2的两种抗体可以同时非竞争性地结合RBD的不同区域,而且病毒同时在两个不同位点发生突变的可能性很小,因此可以降低病毒通过基因突变逃避单抗治疗的可能性。
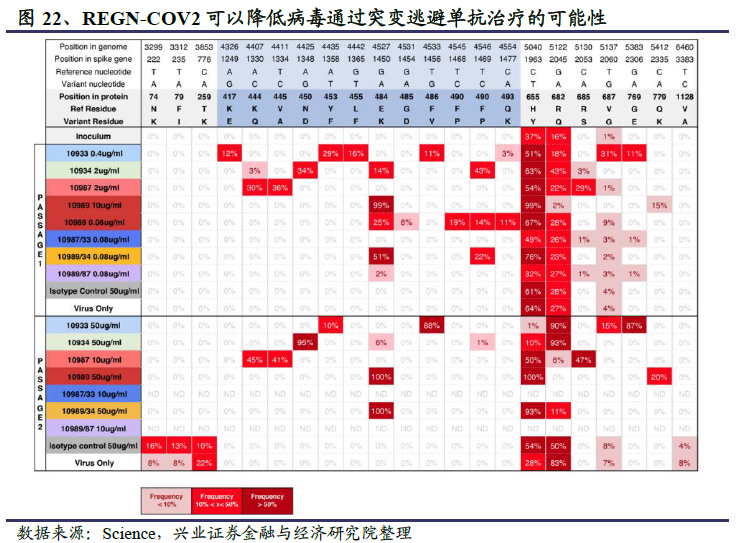
再生元鸡尾酒抗体药物REGN-COV2已经进入预防卫生事件感染的III期临床研究和治疗卫生事件患者的II/III期临床研究。2020年6月11日,再生元的鸡尾酒抗体药物REGN-COV2首次进入临床研究,采用适应性临床设计开启治疗住院以及非住院卫生事件患者的2项临床研究,目前该2项临床研究已经进入II/III期临床。在I期临床试验中,对包含30例住院和非住院卫生事件患者的初始队列的审查发现REGN-COV2表现出良好的安全性;基于该结果,REGN-COV2此次直接进入了预防卫生事件感染的3期临床研究。
2020年7月6日,再生元的鸡尾酒抗体药物REGN-COV2进入预防试验III期临床研究,预防处于高暴露风险的未感染者(例如医护人员或急救人员)和与COVID-19患者密切接触的未感染者(例如患者的家人),这是全球首个进入3期临床的预防公共卫生事件的病毒感染的抗体。此次启动的III期预防试验将在大约100家研究中心进行,预计在美国将招募2000名受试者;该试验将评估SARS-CoV-2感染状态。
在住院(估计入组人数=1850)和非住院(估计入组人数=1050)患者中进行的II/III期治疗试验计划在美国、巴西、墨西哥和智利的约150家研究中心进行,并将评价病毒学和临床终点,预计在今年8月份获得初步数据。所有试验均为适应性设计,入组患者的最终数量将取决于试验进展和II期研究的结果。
2020年7月7日,再生元宣布与美国生物医学高级研究与开发局(BARDA)和美国国防部签订REGN-COV2鸡尾酒抗体的制造和供应协议。根据协议,美国政府将花费4.5亿美元购买相当于7-30万治疗剂量,或42-130万预防剂量的REGN-COV2鸡尾酒抗体药物,具体给药剂量将在临床试验中进行评估并加以确定。再生元预计可在夏末生产出第一批药物,秋季完成全部的药物生产。
2020年8月19日,再生元与罗氏共同宣布达成卫生事件中和抗体药物的生产合作。罗氏拥有全球最大的抗体药物生产基地,与罗氏合作后,再生元生产抗体药物的能力将至少提高3.5倍。两家公司将合作开发和制造鸡尾酒抗体药物REGN-COV2,再生元负责REGN-COV2在美国的销售,而罗氏将负责该药物在全球范围内(美国以外)的分销。
2020年9月14日,再生元与牛津大学宣布将开展一项3期临床试验RECOVERY (Randomised Evaluation of COVid-19 thERapY),以评估再生元鸡尾酒疗法REGN-COV2在2000名英国住院患者中的治疗效果,主要评估REGN-COV2对患者死亡率,住院时间和通风需求的影响。
6.1.3、盈利与估值
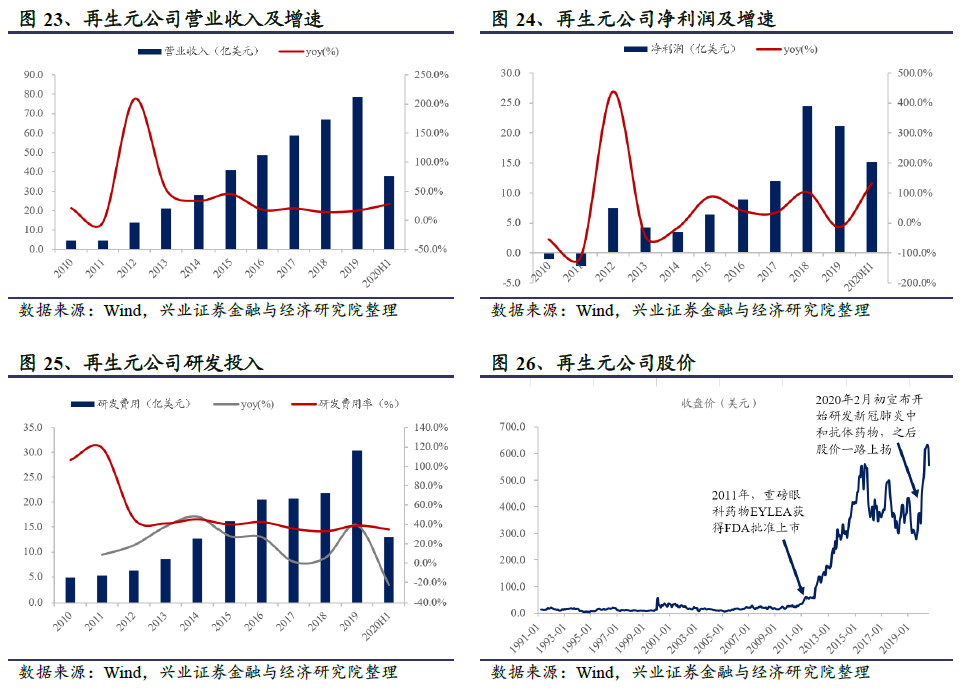
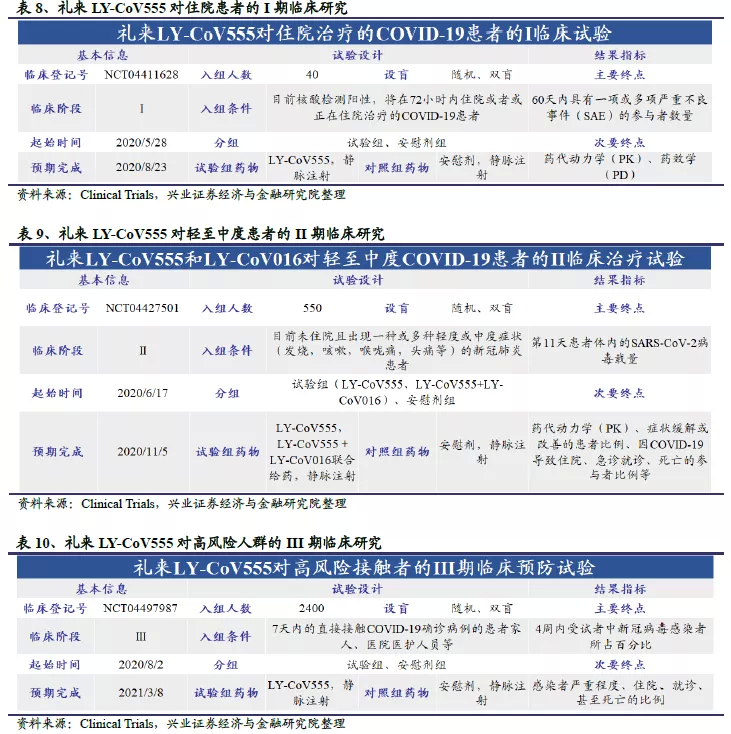
再生元2019年全年实现营业收入78.63亿美元;2020 H1实现营业收入37.80亿美元,同比增长28.13%。公司2019年全年净利润为21.16亿美元;2020 H1实现净利润15.22亿美元,同比猛增132.64%。受公共卫生事件影响,许多制药公司今年上半年业绩出现下滑,而再生元却实现了营业收入与净利润双增长,主要是核心产品眼科药EYLEA、抗炎药Dupixent和PD-1单抗Libtayo的业绩增长,以及美国政府对其抗体鸡尾酒REGN-COV2的订货协议带来的。公司2019年的研发投入为30.37亿美元,占营业收入之比为38.62%,同比增长38.93%。再生元一直非常重视研发投入,是全球制药企业50强中研发费用率最高的公司;2010-2019年研发费用的CAGR为22.5%,研发费用率稳定在40%左右。
公司股价从2011年重磅眼科药物EYLEA获批上市后开始快速上涨,截至2019年12月31日,公司市值414.1亿美元,PE(TTM)19.2。2020年初受公共卫生事件等影响美股大跌,但再生元公司却逆流而上;后虽有回调,截至9月18日股价涨幅仍高达48.7%。同期纳斯达克指数仅上涨18.7%,可见大家对再生元开发的抗体鸡尾酒疗法寄望颇高。截至2020年9月18日,公司市值590.7亿美元,PE(TTM)19.8。
6.2、礼来:通过合作开发寻公共卫生事件的病毒中和抗体突破
礼来公司(Eli Lilly and Company)成立于1876年,由药学家Eli Lilly创建,并最终以他的姓名命名。礼来总部设在美国印第安纳波利斯,拥有全职雇员3.4万名。礼来是一家市值超千亿美元的全球性制药公司,也是全球第一个上市胰岛素和生产抗生素的企业,形成了以中枢神经系统、抗肿瘤、内分泌系统、抗感染、骨质疏松、心血管疾病、糖尿病和免疫学等为主的治疗领域。目前,礼来公司在波多黎各及17个其他国家设有办公机构,在全球超过55个国家/地区进行临床研究,产品销往约125个国家。
6.2.1、中和抗体项目
礼来在卫生事件中和抗体领域主要有两项布局:一是和加拿大生物技术公司AbCellera合作开发了LY-CoV555,二是投资最高2.55亿美元购买君实生物JS016大中华以外地区的临床开发和商业化权益。其中LY-CoV555于6月1日宣布进入I期临床,与君实生物合作开发的JS016于6月7日获批进入I期临床,分别是全球第一、第二个进入临床试验阶段的潜在中和抗体。目前来看,礼来参与的两个中和抗体项目最先进入I期临床试验,或成卫生事件中和抗体研发的最大赢家。JS016的研发及临床进展将在君实生物部分详细介绍,这里重点介绍礼来和AbCellera合作开发的LY-CoV555。
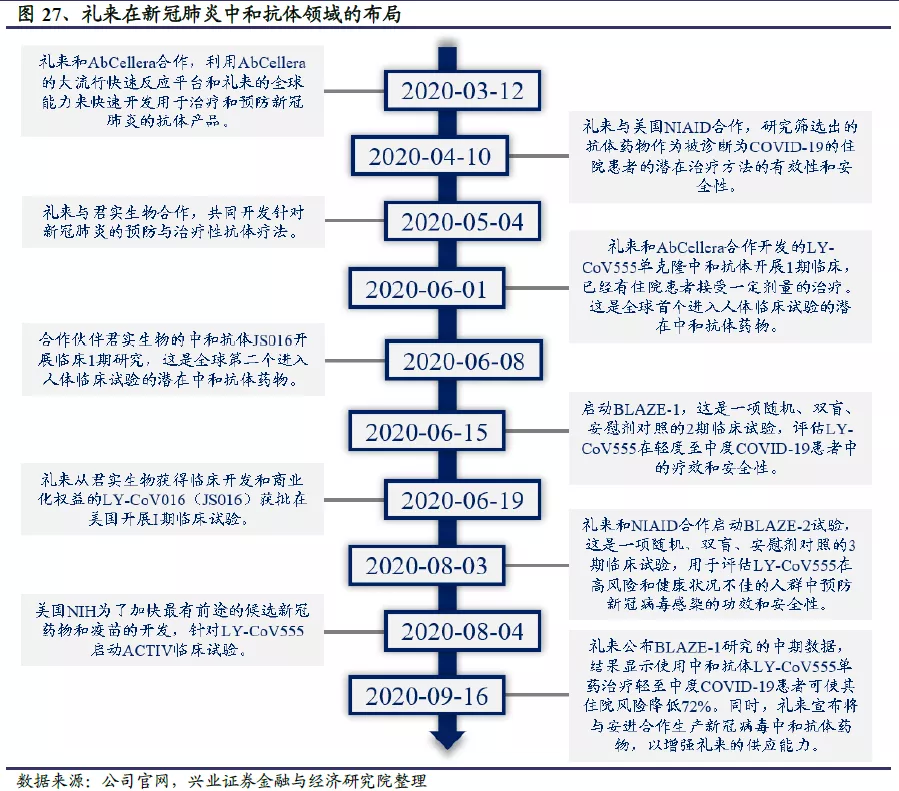
LY-CoV555是由礼来与加拿大生物技术公司AbCellera合作开发的一个潜在用于预防和治疗卫生事件的单克隆中和抗体。在AbCellera和美国国家过敏和传染病研究所(NIAID)的疫苗研究中心从美国首批COVID-19康复者的血样中分离出抗体后,礼来公司的科学家用了不到三个月的时间开发出这种对公共卫生事件的病毒S蛋白具有强效中和作用的单克隆抗体。
2020年6月2日,礼来宣布其与AbCellera合作开发的中和抗体LY-CoV555的1期临床研究(NCT04411628)已对首批患者进行了给药。与安慰剂的对照研究将评估COVID-19住院患者的安全性和耐受性,6月底可得出结果。
I期临床试验的结果表明LY-CoV555在所有测试剂量下均具有良好的耐受性,未观察到与药物相关的严重不良事件(SAE),因此礼来开展了另外2项研究。一项是涉及550例患者的II期临床研究BLAZE-1(NCT04427501),研究对象是最近被诊断出COVID-19的轻至中度患者。礼来预计将于9月完成BLAZE-1的入组,此后会很快公布初步结果,并在2020年11月获得完整数据。另一项III期临床试验BLAZE-2将招募2400名受试者,该临床试验将在美国政府的帮助下,在有感染高风险和健康状况不佳的人群——长期护理机构(通常指疗养院和辅助生活社区)的居住者和工作人员中进行,测试单剂量LY-CoV555是否能在4周内预防公共卫生事件的病毒感染,以及是否能在8周内减少COVID-19并发症。礼来公司发言人表示,II期和III期试验的数据披露高度依赖于患者入组。如果LY-CoV555被证明有效,到今年年底将有10万剂以上的药物可用。
2020年8月4日,美国国立卫生研究院NIH启动加速计划ACTIV,这是一项美国政府联合企业开展的公私合作计划,旨在加速有潜力的公共卫生事件的病毒候选药物和疫苗的研发。该计划针对礼来的LY-CoV555开展ACTIV-2和ACTIV-3两项研究,ACTIV-2针对轻至中度患者,目前在开展II期临床试验,主要目标是评估抗体药物的安全性及有效性。如果药物没有严重的安全隐患,并且疗效符合标准,将进入III期临床试验。ACTIV-3是针对住院患者的III期临床试验,主要目的与ACTIV-2相同。据悉,LY-CoV555是该试验评估的第一个药物,稍后可能还会在同一试验方案下研究其他治疗药物。
2020年9月16日,礼来公布BLAZE-1研究的中期结果,显示使用中和抗体LY-CoV555单药治疗轻至中度COVID-19患者,可使其住院率降低72%。BLAZE-1研究针对新确诊的轻至中度COVID-19患者,计划入组800例受试者,分别进行LY-CoV555单药治疗和LY-CoV555+LY-CoV016联合给药治疗。其中LY-CoV555单药治疗组入组452例患者,分别给予安慰剂和700mg、2800mg和7000mg的LY-CoV555。主要终点是第11天患者体内病毒载量较基线水平的变化,次要终点还包括安全性、第29天因COVID-19导致住院、急诊或死亡的受试者比例等。结果显示,2800mg剂量水平LY-CoV555治疗达到了预设的主要终点,其他剂量组均未达到主要终点;但大多数患者,包括接受安慰剂的患者,在第11天都能达到病毒几乎完全清除的状态。合并各剂量组数据综合分析后发现,LY-CoV555治疗患者中住院或急诊的发生率为1.7%(5/302),安慰剂组为6%(9/150),提示LY-CoV555可使轻至中度COVID-19患者的住院和急诊风险降低72%。此外,LY-CoV555耐受性良好,无药物相关严重不良事件报道。各剂量组的治疗相关紧急不良事件与安慰剂组相似。病毒RNA测序显示,安慰剂组和各剂量治疗组均有发生LY-CoV555耐药变异,LY-CoV555治疗组的耐药发生率比安慰剂组要高(8% vs 6%)。
同日,礼来和安进宣布将合作生产公共卫生事件的病毒中和抗体药物,以增强礼来的供应能力。两家公司预计2021年将生产百万剂中和抗体药物。
6.2.2、盈利与估值
礼来2019年全年实现营业收入223.20亿美元;2020 H1实现营业收入113.59亿美元,同比增长5.87%。近几年公司营业收入已趋于稳定,2015-2019年营收CAGR为2.8%。公司2019年全年净利润为83.18亿美元;2020 H1实现净利润28.69亿美元,同比下滑48.49%。公司2020 H1研发投入为27.82亿美元,占营业收入之比为24.49%。最近五年礼来研发投入稳中有增,CAGR为3.9%,费用率维持在20-25%之间。
1999-2007年间,由于礼来在百忧解等多种重磅产品专利断崖后青黄不接,营收和净利润快速下滑,股价也随之一跌再跌。之后礼来多方寻求合益的合作模式,以便尽快获得具有潜力的药品管线和技术,比如和信达生物合作开发以PD1为代表的药品,此次卫生事件购买君实生物JS016大中华区以外的临床开发和商业化权益等。实践证明礼来这条路线取得了成功,公司股价在2010年开始稳步回升,2018年后开始快速上涨,截至2019年12月31日,公司市值1259.2亿美元,PE(TTM) 15.9。2020年初受公共卫生事件等影响美股大跌,公司股价受到一定影响后迅速反弹,截至2020年9月18日,市值1474.6亿美元,PE(TTM)26.2。
6.3、Vir Biotechnology:与GSK合作开发公共卫生事件的病毒单克隆中和抗体
Vir Biotechnology成立于2016年,2019年10月在纳斯达克上市,总部位于美国加州旧金山,拥有全职雇员220余人。VIR是一家临床阶段的免疫学公司,致力于将免疫学见解与尖端技术相结合,以治疗和预防严重的传染病,其使命是创建一个没有传染病的世界。
6.3.1、技术平台
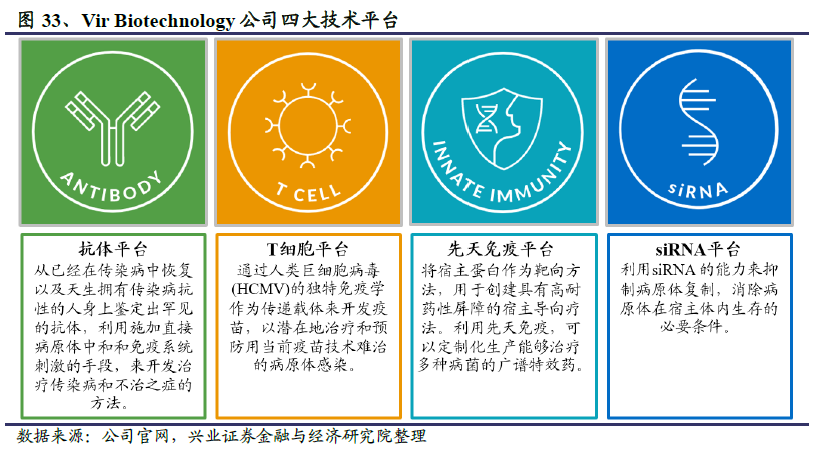
VIR认为应用免疫学的最新进展来对抗传染病的时代已经来临。为此,VIR采用的方法是先确定免疫系统在对抗特定病原体方面的局限性、该病原体的脆弱性以及先前方法失败的原因;然后利用强大的技术平台(这些技术可以单独或组合使用)开发有效的治疗方法。VIR通过内部开发、协作和收购组建了四大技术平台:抗体平台、T细胞平台、先天免疫平台和siRNA 平台。其中抗体平台用于识别和开发针对病原体的抗体药物,覆盖的病原体包括乙肝病毒(HBV),甲流病毒和公共卫生事件的病毒等。VIR利用该抗体平台对发现的全人源抗体进行工程改造,以增强其治疗潜力。
6.3.2、研发管线
目前VIR的的研发管线主要针对乙型肝炎(HBV)、甲型流感、卫生事件、艾滋病(HIV)和肺结核(TB)等,单独或组合使用公司现有技术平台开发有效疗法。除了针对公共卫生事件的病毒SARS-CoV-2的中和抗体药物外,公司针对乙肝病毒HBV、甲流病毒也有开发单克隆中和抗体药物。
1)乙肝
全球每年约有2.9亿人长期感染乙肝病毒HBV,其中约90万人死于相关并发症。VIR-2218是皮下注射的靶向HBV的siRNA药物,VIR-3434是皮下注射的HBV单克隆中和抗体。VIR-2218和VIR-3434有望带来乙肝的功能性治愈,意味着在有限的治疗时间后能够终身控制病毒的复制。
2)甲型流感
VIR-2482是一种预防甲型流感的单克隆中和抗体,2019年8月开始进行I期临床试验,临床前试验显示VIR-2482已涵盖自1918年西班牙流感大流行以来出现的所有主要甲型流感病毒株,半衰期持续五到六个月,可覆盖整个流感季。
3)卫生事件
VIR-7831和VIR-7832都是单克隆中和抗体,已被证明具有体外中和SARS-CoV-2病毒的能力。这两种抗体与SARS-CoV-2病毒上和SARS-CoV病毒的共享表位结合,研究表明该表位高度保守,产生突变逃避抗体治疗的可能性更小。VIR-7831和 VIR-7832已经过精心设计,延长了半衰期;其中VIR-7832还可以充当T细胞疫苗。
6.3.3、中和抗体项目
2020年5月,VIR在Nature上发表研究,对从SARS康复患者体内分离的S309抗体进行了详细的鉴定和表征。以往的临床试验证明单克隆中和抗体能降低埃博拉病人病毒水平,起到有效中和病毒毒力、实质性改善临床症状、降低感染者死亡率等作用。治疗埃博拉病毒感染的单克隆中和抗体mAb114已被美国FDA授予突破性疗法和孤儿药资格。VIR正在使用与开发mAb114相同的平台开发S309抗体以治疗卫生事件。基于S309抗体,VIR和葛兰素史克(GSK)合作开发了两种候选单克隆抗体药物VIR-7831和VIR-7832。
VIR-7831和VIR-7832能够有效中和公共卫生事件的病毒,且能够有效防止公共卫生事件的病毒因突变产生的抗药性。具体而言,二者靶向SARS病毒(SARS-CoV)和公共卫生事件的病毒的共享表位,该表位高度保守,研究证明经过10次传代后没有发生突变逃逸抗体治疗。两种单克隆抗体在体外都表现出免疫效应,允许招募免疫细胞来杀死已经感染的细胞。两种抗体的Fc片段已经过工程化改造,以提高患者肺部的生物利用度和延长半衰期;Fc区域经过“XX2”突变改造的VIR-7832还可以充当T细胞疫苗。
2020年8月31日,VIR和GSK宣布上周完成了合作开发的公共卫生事件的病毒中和抗体VIR-7831(也称为GSK4182136)2/3期临床试验中第一例患者的给药。这项名为COMET-ICE的研究将在全球范围内招募约1300名卫生事件早期感染者,目的是评估单剂量的VIR-7831用于预防轻至中度COVID-19患者住院的安全性和有效性。研究预计在今年年底前获得初步结果,在2021年第一季度获得完整结果,并尽可能在2021年上半年让患者接受该抗体药物的治疗。未来VIR和GSK还将针对VIR-7831开展另外两项临床试验,一项用于治疗重症住院患者,另一项用于预防公共卫生事件的病毒感染。两家公司预计在今年晚些时候开展另一个在研中和抗VIR-7832的2期临床试验,该抗体具有与VIR-7831相同的特征,同时可以作为治疗和/或预防性T细胞疫苗。
此前VIR在公司公告中表示,如果这些中和抗体药物被证明有效,预计2021年将生产1000万-1500万剂,其中药明生物负责临床试验所需的初始生产,Biogen和三星生物负责大规模商业化生产。VIR和GSK关于SARS-CoV-2中和抗体的合作项目分工明确,VIR负责研发,GSK主导商业化,两家公司各自负责部分临床生产,销售收入将按照VIR75%、GSK25%的比例进行分配。VIR和GSK在开发公共卫生事件的病毒治疗药物方面开展了多项合作,除了中和抗体药物,还包括疫苗和基因治疗药物。
6.3.4、盈利与估值
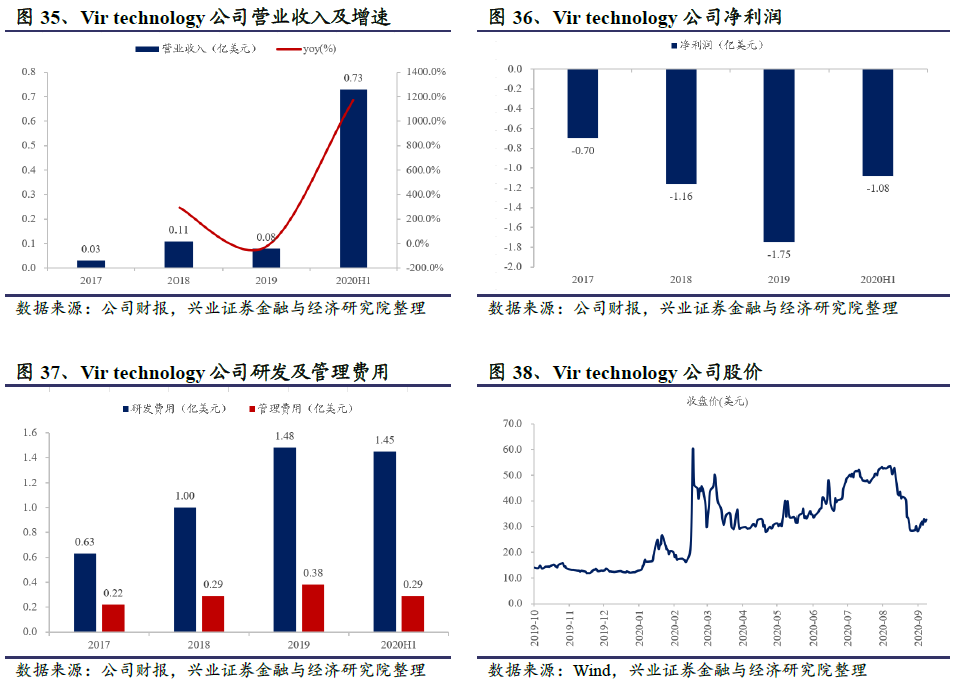
VirBiotechnology 暂无产品获批上市,主要靠非核心业务创收,收入较低且波动较大。2019年全年实现营业收入809万美元;2020 H1实现营业收入7271万美元,同比增长1173.8%。公司2019年全年继续亏损1.75亿美元,2020 H1亏损1.08亿美元。2017-2020 H1公司研发费用分别为0.63、1.00、1.48和1.45亿美元,主要原因是公司处于生物创新药行业,多个在研产品进入临床临床前试验阶段,导致公司研发费用持续增长。
公司于2019年10月在纳斯达克上市,发行价20美元/股,上市当天破发近30%。此后股价一路走低,截至2019年12月31日,公司市值13.5亿美元,PS(TTM) 134.7。2020年2月公司宣布研发卫生事件中和抗体药物后,股价一度飙升至60.2美元/股,之后快速回调;4月公司宣布GSK投资2.5亿美元共同开发单克隆中和抗体后,股价再次上扬;9月18日回调至32.7美元/股。截至2020年9月18日,公司市值41.6亿美元,PS(TTM) 55.4。
6.4、君实生物:JS016国内领跑,与礼来达成合作协议
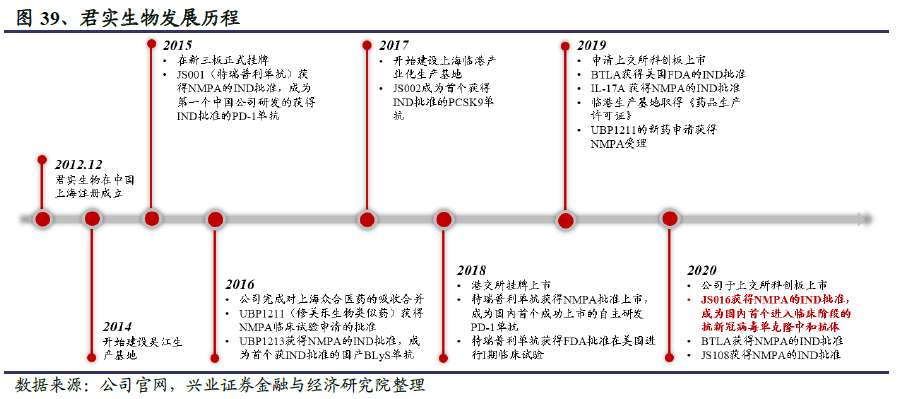
君实生物成立于2012年12月,是一家以开发治疗性抗体为主的创新驱动型生物制药公司,致力于创新药物的发现和开发,以及在全球范围内的临床研发及商业化。公司目前自主研发的重磅PD-1单抗(特瑞普利单抗)已上市销售,在研管线丰富,主要覆盖肿瘤、代谢、自身免疫、神经系统、抗感染等重大疾病领域。
公司旨在通过源头创新来开发首创(First-in-class)或同类最优(Best-in-class)的药物。在中国区域内,君实生物凭借卓越的创新药物发现能力、先进的生物技术研发、全产业链大规模生产技术及快速扩大极具市场潜力的在研药品组合,在肿瘤免疫疗法、自身免疫性疾病及代谢疾病治疗方面处于领先地位。公司成立以来发展迅速,已成为国内首家获得抗PD-1单克隆抗体NMPA上市批准、抗PCSK9单克隆抗体NMPA临床申请批准的中国公司,并取得了全球首个治疗肿瘤抗BTLA阻断抗体在中国NMPA和美国FDA的临床申请批准。
6.4.1、技术平台
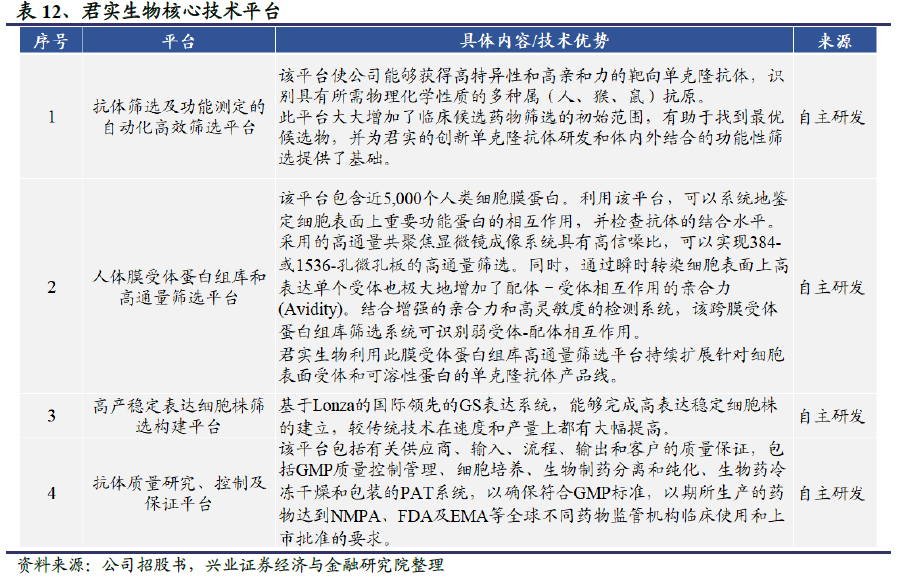
君实生物已自主研发建立了涵盖蛋白药物从早期研发到产业化的整个生命周期的完整技术体系,包括七个主要的技术平台:(1)抗体筛选及功能测定的自动化高效筛选平台,(2)人体膜受体蛋白组库和高通量筛选平台,(3)抗体人源化及构建平台,(4)高产稳定表达细胞株筛选构建平台,(5)CHO 细胞发酵工艺开发平台,(6)抗体纯化工艺及制剂工艺开发与配方优化平台,(7)抗体质量研究、控制及保证平台。其中,抗体筛选及功能测定的自动化高效筛选平台、人体膜受体蛋白组库和高通量筛选平台、高产稳定表达细胞株筛选构建平台和抗体质量研究、控制及保证平台为公司的核心技术平台。
6.4.2、中和抗体项目
从进度上看,从中科院微生物所2月初从康复期病人内筛选出全人源单克隆中和抗体到6月初进入临床试验,君实生物JS016单克隆中和抗体的临床前研发仅用时4个月。JS016是国内首个、全球第二个进入临床人体试验阶段的潜在中和抗体药物,7月7日已完成受试者给药,预计9月能获得初步结果。
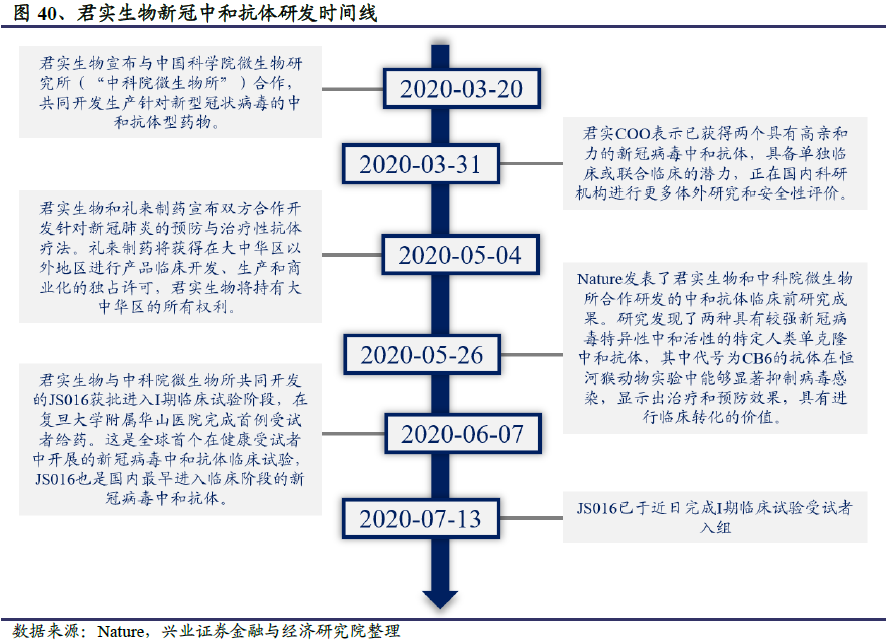
2020年1月中旬,中科院微生物所组建研究团队,从康复病人血清体内分离鉴定到几十株全人源抗体。
2020年3月20日,君实生物宣布与中科院微生物所合作,共同开发生产中和抗体。君实生物与中科院微生物所发挥各自优势,联合攻关,目前已获得多株能够阻断病毒入侵的中和抗体并开展了动物实验,初步的体外实验验证了抗体的阻断活性。
2020年5月4日,君实生物宣布与礼来合作研发卫生事件中和抗体药物。礼来将获得大中华区以外地区进行产品临床开发、生产和商业化的独占许可,君实生物将持有大中华区的所有权利。礼来表示将在今年第二季度向美国FDA申报其在美的临床试验。合作协议的具体内容包括:(1)授权礼来在大中华区以外对君实卫生事件抗体开展研发、生产和销售的独占许可;(2)礼来将向公司支付1000万美元首付款,并在君实每个卫生事件抗体(单用或组合)实现规定的里程碑事件后,向公司支付最高2.45 亿美元的里程碑款,外加该产品销售净额两位数百分比的销售分成;(3)双方将推进卫生事件抗体新药临床试验和商业化,按照双方同意的条款和条件下,礼来将以7500万美元认购君实生物新发行的H股股份(潜在认购)。
2020年5月26日,君实生物与微生物所合作研发的中和抗体临床前研究成果发表在Nature上,研究发现了两种具有较强公共卫生事件的病毒SARS-CoV-2特异性中和活性的特定人类单克隆中和抗体,其中代号为CB6的在体外实验中表现出更强的阻断能力,不仅比宿主细胞受体的亲和力高100-200倍以上,且与病毒结合区域高度重叠,使得病毒与宿主细胞结合的可能性大大降低。在恒河猴动物实验中,CB6抗体能够显著抑制病毒感染,显示出治疗和预防效果。
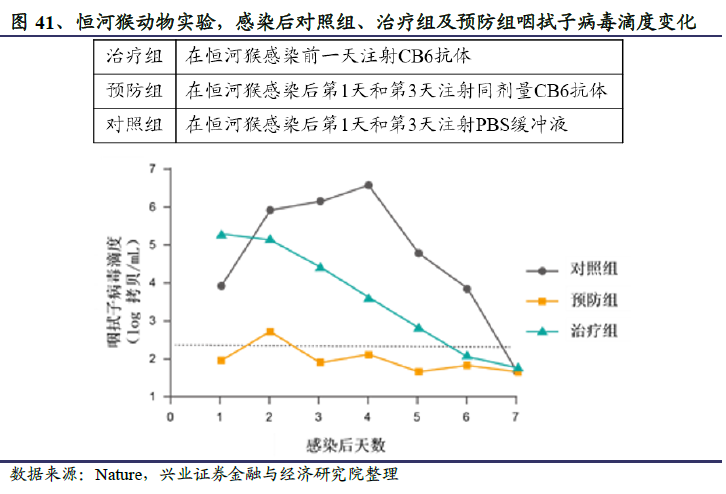
君实生物基于CB6抗体开发了卫生事件中和抗体药物JS016。临床前研究显示,JS016对公共卫生事件的病毒S蛋白的RBD结合域表现出极高的特异性亲和力,达到nM水平,能够抢先与病毒结合,从而从源头阻断病毒入侵宿主细胞。在有效性方面,恒河猴动物感染实验显示JS016表现出治疗和预防效果。这是全球首个报告抗公共卫生事件的病毒中和抗体的非人灵长类动物实验结果。在安全性方面,JS016抗体基因来源于康复病人单个B细胞,是全人源天然抗体,经人体免疫系统筛选,不结合人体自身抗原,预期有较好的安全性。此外,为了减少抗体介导的急性肺损伤风险,确保临床应用中的安全性,研发团队对JS016进行了Fc区域改造,有效降低了可能存在的抗体依赖增强效应(ADE)、抗体介导的细胞毒作用(ADCC)以及细胞吞噬作用(ADCP)。
2020年6月7日,君实生物和中科院微生物所共同开发的重组全人源单克隆抗体JS016获批进入I期临床试验阶段,在复旦大学附属华山医院完成首例受试者给药。JS016是国内最早、全球第二个进入临床阶段的公共卫生事件的病毒中和抗体。
该临床试验是一项随机、双盲、安慰剂对照的I期临床研究,由复旦大学附属华山医院张菁教授与张文宏教授联合主持,旨在评价JS016静脉注射给药在中国健康志愿者中的耐受性、安全性和药代动力学特征及免疫原性,为探索JS016在人体中抗公共卫生事件的病毒的治疗与预防疗效提供依据。
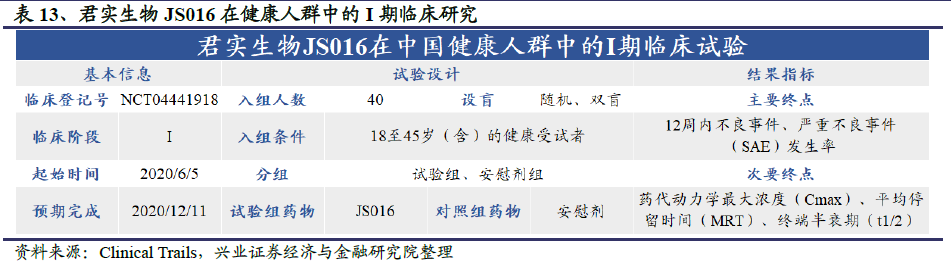
2020年7月7日,国内的临床试验按照既定方案,完成4个剂量组共40例受试者给药,截至7月13日,尚未观察到剂量限制性事件(DLE),为进一步探索JS016新药在人体中抗公共卫生事件的病毒的疗效提供安全性依据。君实生物计划尽快开展针对轻型/普通型卫生事件患者的Ib期临床研究,以及针对重型及危重型卫生事件患者的II/III期临床研究。同时,公司也将后续开展针对公共卫生事件的病毒高危人群的研究,以评价JS016对公共卫生事件的病毒感染的预防作用。
2020年5月礼来与君实生物签订中和抗体合作开发协议后,2020年6月19日,礼来已启动JS016(LY3832479)在美国的I期临床试验,计划在24名健康参与者中以单次静脉注射剂量评估LY3832479的耐受性,安全性,药代动力学和免疫原性。随后,礼来启动LY3832479针对轻至中度COVID-19患者的II期临床治疗实验,评估其与LY-CoV555联合给药时缓解或改善患者临床的情况。
6.4.3、盈利与估值
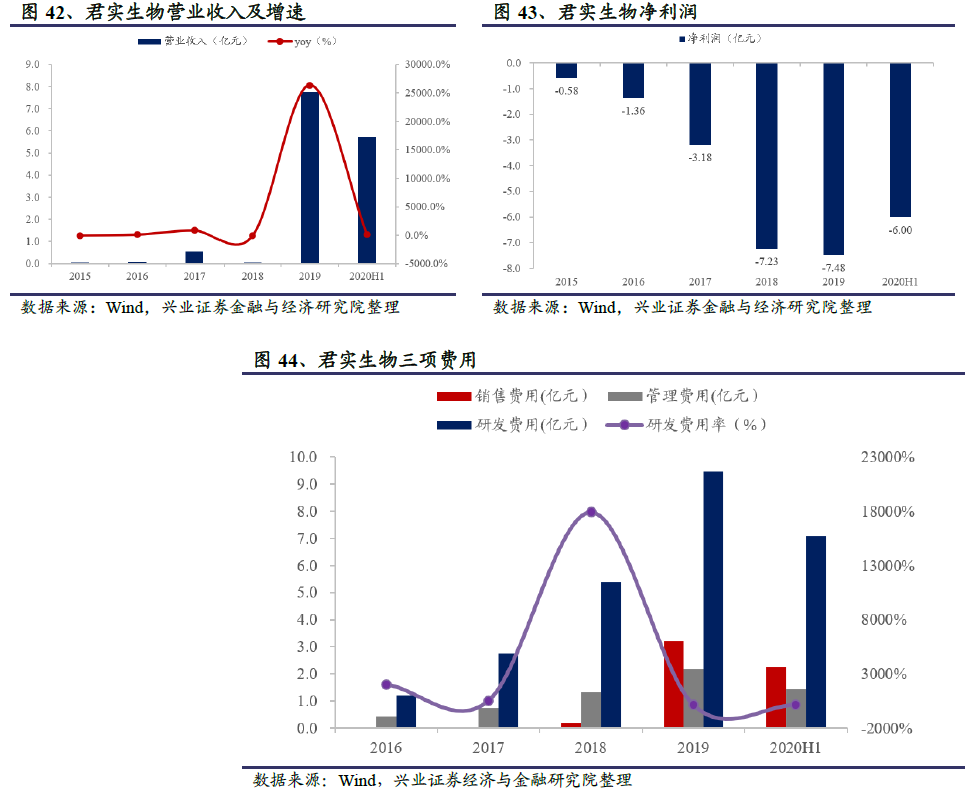
2019年以前公司主要靠非核心业务创收,收入低且波动大;2019年2月首个自主研发的PD-1单抗药物特瑞普利单抗上市后,营业收入开始跨越式提升。2019年君实生物全年实现营业收入7.75亿元,同比猛增26375%;2020 H1实现营业收入5.75亿元,同比增长85.9%。公司2019年全年继续亏损7.48亿元,2020 H1亏损6.00亿元。随着公司上市品种特推普利单抗获批适应症的拓展及阿达木单抗生物类似药等UBP1211获批上市,预计公司营收将进入高速增长期,2022-23年有望实现盈利。2020 H1公司研发、销售和管理费用分别为7.09亿元、2.28亿元和1.44亿元,研发费用为营业收入的123.3%,同比2019 H1的119.4%增长3.9 pp。君实生物一直非常重视研发,研发支出显著高于同行业上市公司平均水平且一直稳步提升。主要原因是公司处于生物创新药行业,多个在研产品进入临床阶段,因而研发费用持续增长。
君实生物2020年7月15日在科创板上市,此前在2018年底已登陆港交所。截至2020年9月18日,公司市值720.5亿元,PS(TTM)77.5。
6.5、神州细胞:国内第四个进入临床的中和抗体
神州细胞是一家创新型生物制药研发公司,主要从事单克隆抗体、重组蛋白和疫苗等生物药的研发生产,专注于恶性肿瘤、自身免疫性疾病、感染性疾病和遗传病等多个治疗和预防领域。集团下属的神州细胞工程有限公司创办于2002年,是中国最早专门从事大分子生物药研发的企业之一。2020年6月,公司在科创板挂牌上市。
6.5.1、技术平台
自成立以来,公司专注于研发具有领先技术水平和成本优势的生物药,建立了生物药上、中、下游全链条技术平台,包括五大核心技术体系:(1)创新中和抗体候选药物发现技术体系;(2)生物药高效生产工艺技术体系;(3)生物药质量控制技术体系;(4)生物药成药性评价技术体系;(5)规模化生产及管理技术体系。
6.5.2、中和抗体项目
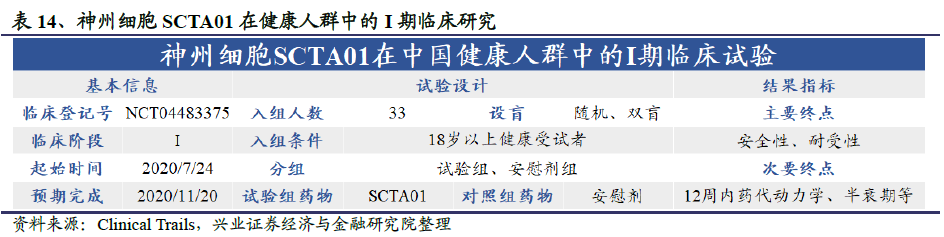
ClinicalTrails资料显示,神州细胞研发的针对公共卫生事件的病毒的中和抗体SCTA01已经于7月24日进入临床试验阶段。这是一项随机、双盲、安慰剂对照、单次剂量研究的I期临床试验,目的是评估健康中国受试者中SCTA01的耐受性,安全性和药代动力学。
6.6、腾盛博药:与清华、深圳三院合作开发公共卫生事件的病毒中和抗体
腾盛博药成立于2018年4月,在中美两地都设有分部,专注于加快创新和优化中国患者获取药物的渠道。同时,公司宣布完成2.6亿美元A轮融资,由ARCH Venture Partners、通和毓承资本、博裕资本、红杉资本中国基金、云锋基金和蓝池资本领投。
6.6.1、中和抗体项目
2020年3月31日,腾盛博药宣布与清华大学、深圳市第三人民医院合作,共同推动针对公共卫生事件全人源单克隆中和抗体的研究、转化、生产和商业化。
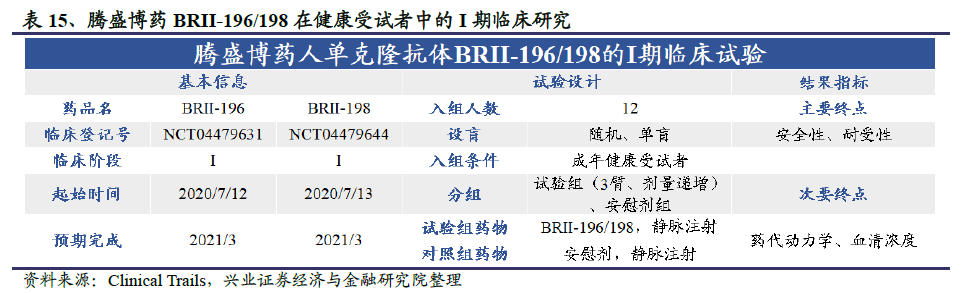
清华大学张林琦教授实验室与深圳市第三人民医院张政团队在国内康复患者中筛选出多个具有治疗潜力的全人源单克隆中和抗体(mAb)。基于此项研究,三方将共同推进多个候选分子项目以用于公共卫生事件的病毒感染的预防和治疗。通过合作,他们在6个月内完成了从选定先导药物到在人体中进行首剂用药的临床试验。目前基于筛选出的单克隆中和抗体开发的两款候选药物BRII-196和BRII-198已进入I期临床试验,主要目的是评估安全性、耐受性和药代动力学。
小结
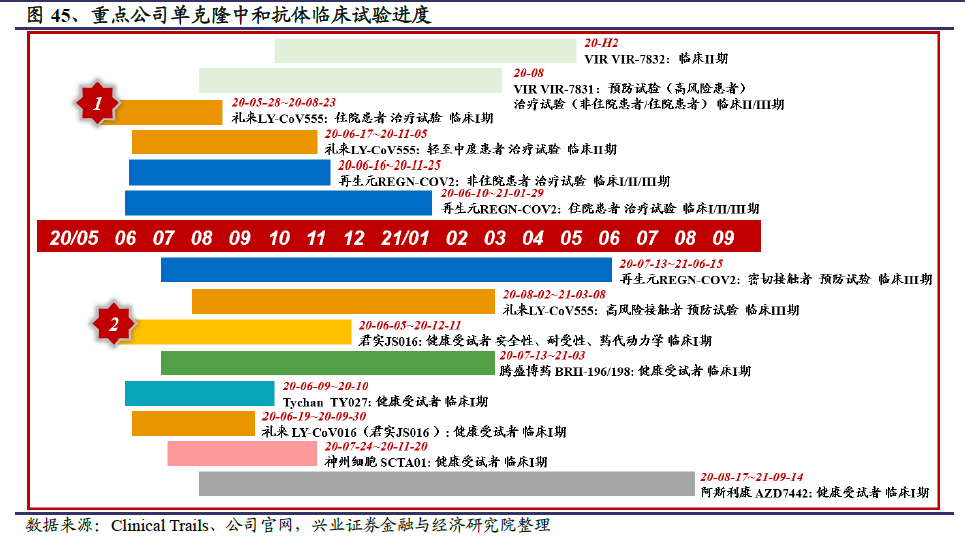
目前SARS-CoV-2中和抗体进入临床试验阶段的海外公司主要有礼来、再生元、Vir Biotechnology和阿斯利康等,国内企业中君实生物领跑。从临床试验的阶段来看,大多数中和抗体候选药物集中在临床I期试验,目的是评估候选抗体的安全性、耐受性和药代动力学。礼来×AbCellera的LY-CoV555和再生元的REGN-COV2进度最快,已经启动旨在预防卫生事件的III期临床试验。其中,礼来LY-CoV555针对高风险人群的暴露前预防,入组的受试者主要是疗养院和辅助生活社区的居住者和工作人员等;再生元REGN-COV2的预防试验覆盖全面,入组的受试者为卫生事件确诊病例的家庭密切接触者(暴露后预防)和医护人员及急救人员(暴露前预防)。礼来和再生元的能够快速推进临床试验的原因是,他们在获得I期临床的初步结果后就会迅速开展II/III期临床,然后多项临床试验并行,抢占研发赛道先机。
从进入临床试验的时间来看,礼来LY-CoV555是全球最先进入临床试验的单克隆中和抗体,6月底已证明具有良好的安全性和耐受性。君实生物的JS-016是全球第二个、中国第一个进入临床试验的中和抗体,礼来买断大中华区以外的开发和商业化权益后,也于6月19日启动了在美国的临床试验。目前国内进入临床试验的中和抗体还有腾盛博药的BRII-196/198、迈威生物的MW33和神州细胞的SCTA01。
从临床试验的揭盲时间来看,进展最快的再生元和礼来均布局了预防卫生事件的III期临床试验,预计1-2个月内即可获得初步的统计学结果。治疗卫生事件方面,再生元针对住院和非住院患者的2项临床研究已经进入II/III期临床,预计9月底将得到III期临床试验的初步数据。礼来预计9月完成轻至中度患者II期临床治疗试验的入组,9月16日已公布中期试验结果。然后将迅速推进对于轻中度或重度住院患者的另外两项III期临床研究,预计年底前拿到III期临床试验的初步结果。VIR和GSK针对VIR-7831开展的II/III期临床试验也预计年底前获得III期临床试验的初步结果。君实生物JS016在国内的I期临床试验已于7月7日完成受试者给药,预计9月获得初步结果。
在中和抗体药物的研发方面,再生元抗体鸡尾酒REGN-COV2在临床试验上领先,礼来的LY-CoV555也紧随其后,国内企业中君实生物走在前列。因而我们预计再生元或礼来有望成为全球率先跑到中和抗体研发赛道终点的公司,君实生物将成为国内首个成功研发公共卫生事件的病毒单克隆中和抗体的企业。
风险提示
1、公共卫生事件提前结束或疫苗先行研发成功的风险:中和抗体主要用于卫生事件的预防及治疗,如果群体普遍接种疫苗或公共卫生事件突然结束,将对中和抗体药物的需求产生不利影响;
2、中和抗体研发失败的风险:中和抗体研发项目具有技术要求高、研发投入大的特点,若临床研发失败将会影响产品上市进度,甚至无法上市,对公司盈利产生不利影响;
3、竞争格局加剧的风险:中和抗体研发处于快速发展及阶段,随着更多企业的加入和疫苗研发的加快,行业竞争程度将更加剧烈。
(编辑:张金亮)




