

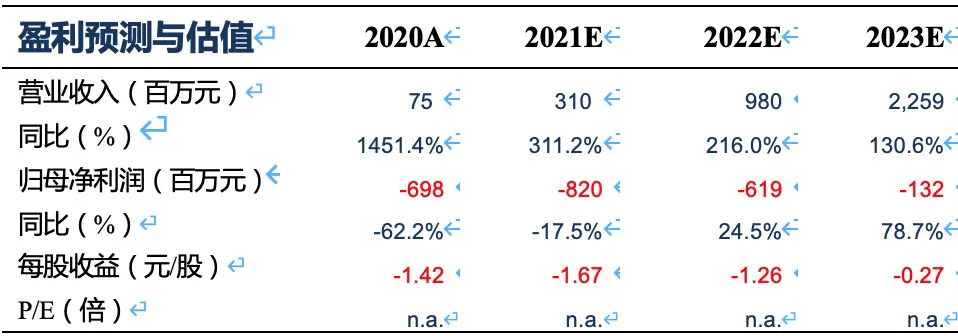
投资要点
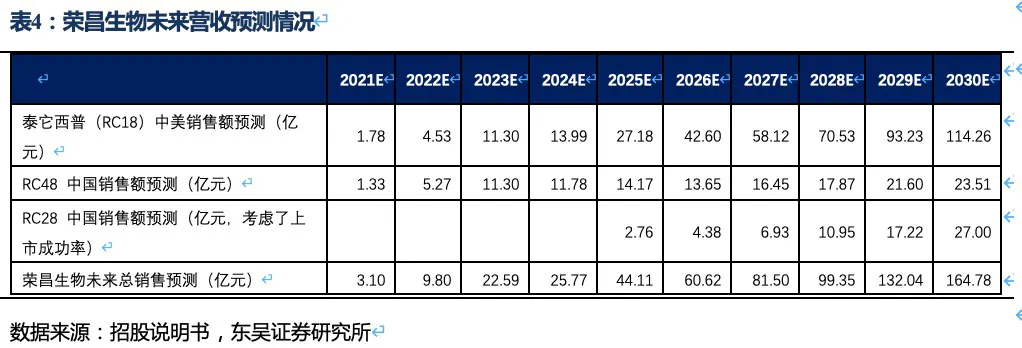
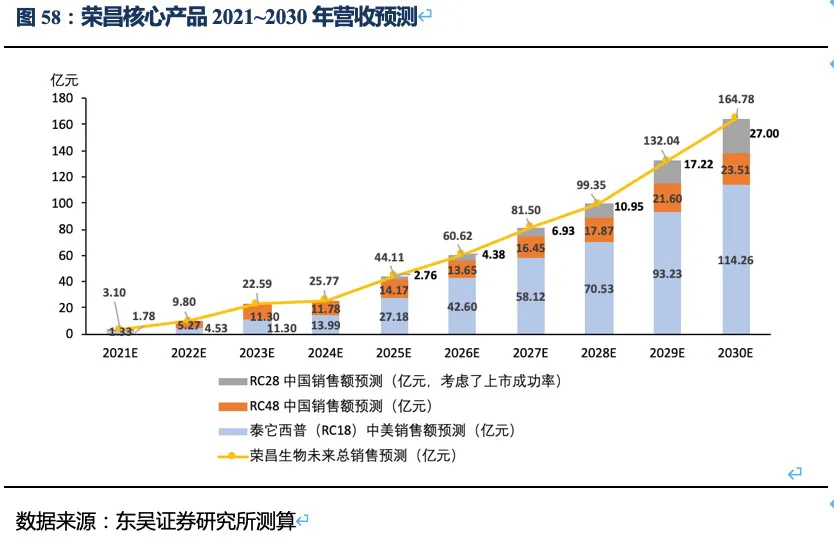
泰它西普相比安慰剂在系统性红斑狼疮(SLE)展现出极好的治疗效果(临床应答率:79.2% VS 32%),成为全球首个双靶点风湿免疫生物创新药,针对SLE适应症在今年3月国内获批上市,另有6个适应症处于III期临床或注册性II期临床阶段。6个核心适应症在国内外的竞争格局良好,尤其在SLE领域仅有GSK的贝利尤单抗唯一竞品,且疗效远好于后者;在IgA肾炎和干燥综合征领域的研发进度也居全球领先地位,且近5年内都不会出现竞品。同时泰它西普在美国开展SLE的III期临床和IgA肾炎的II期临床。鉴于国内风湿免疫市场生物药占比远低于世界平均水平(28.6% VS 68.4%),泰它西普在国内外的上百亿销售潜力行则将至,我们预计泰它西普2021~2023年国内销售额分别为1.78、4.53和11.30亿,2030年其国内外销售额合计超过114亿人民币。
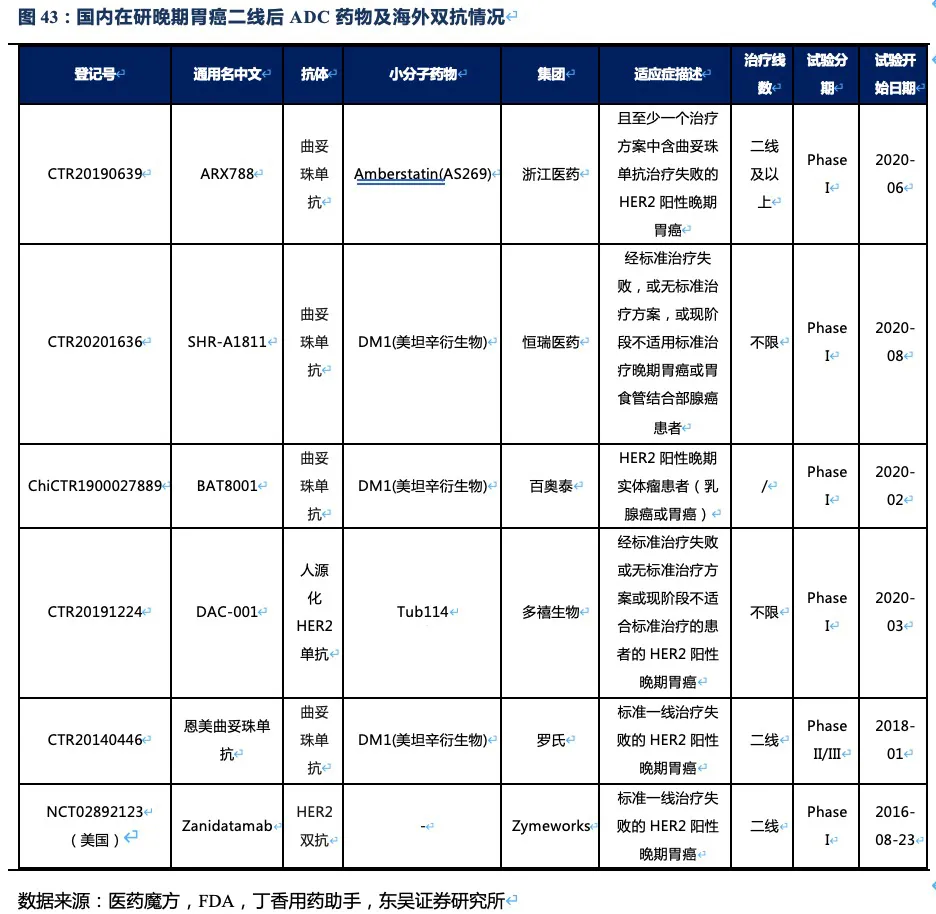
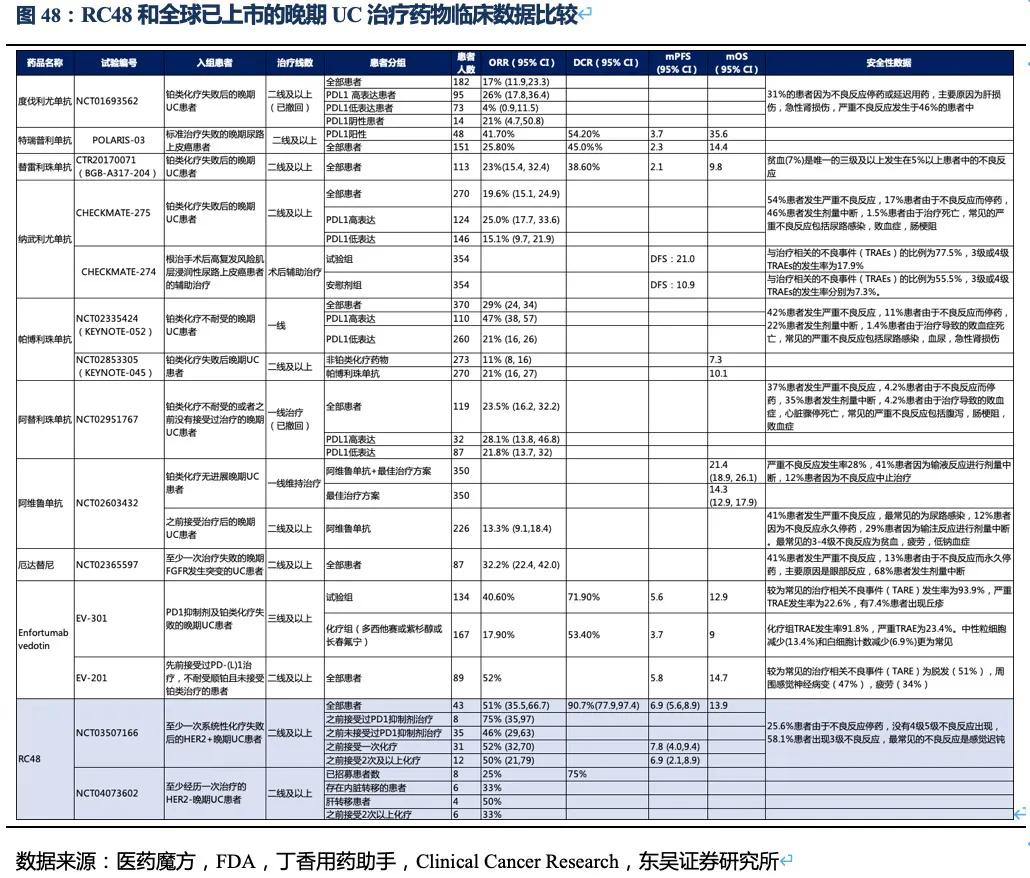
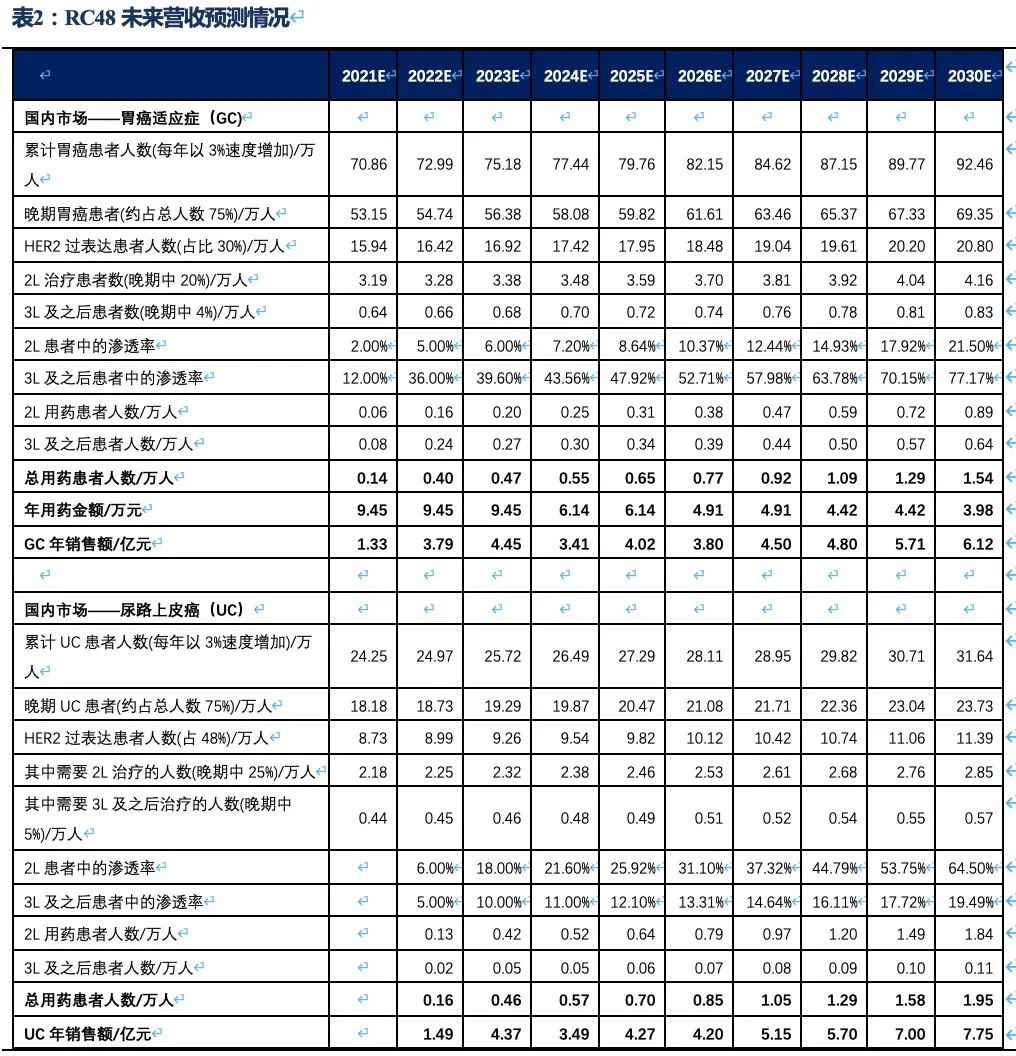
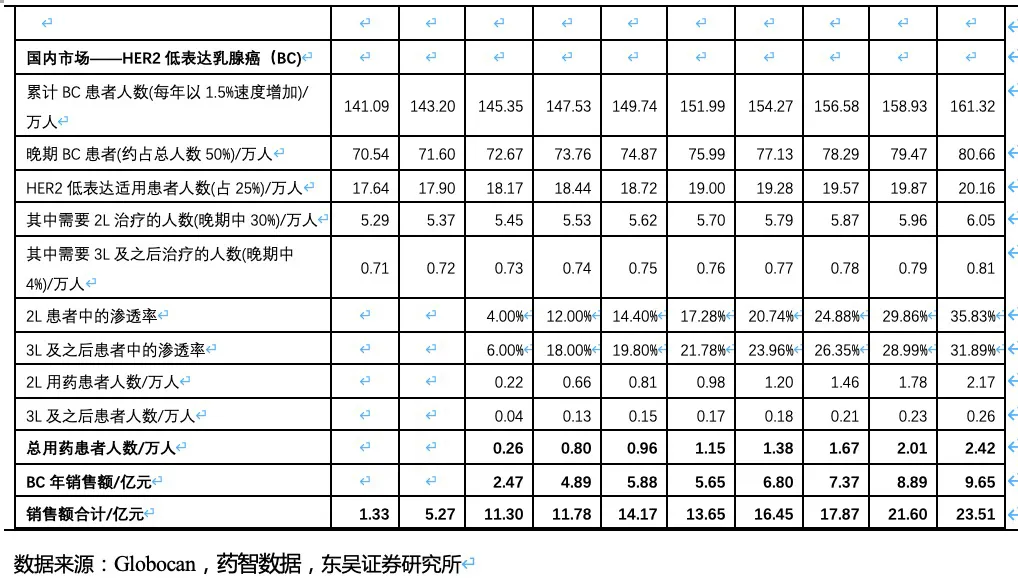
RC48-ADC在Her2过表达晚期胃癌及尿路上皮癌中达到24.4%、60.5%的ORR。随着RC48以II期临床数据在今年6~7月国内获批上市,RC48将会成为胃癌三线的最佳治疗药物,后续也将成为UC二线治疗中目前全球最好的治疗药物。市场认为RC48在胃癌适应症的数据较DS-8201差很多,实际上无论ORR还是OS数据的差异更多是入组患者特征的差异较大导致的。而且DS-8201对于HER2低表达胃癌的亚组数据分析结果也并非优于RC48。相对DS-8201、ARX788、A166三种ADC药物,RC48展现出更好的安全性(无间质性肺炎或眼毒性产生),治疗线数前移的潜力很大,后续可能开展与PD-1单抗的联合试验。此外,RC48对于HER2低表达实体瘤患者也有疗效,目前也在开展针对HER2低表达的胃癌(I期)、尿路上皮癌(II期)和乳腺癌(III期)的临床试验,目标患者数将大幅提高。若仅测算国内市场三个适应症的空间,我们预计2030年RC48将在国内实现近24亿人民币的销售额。
作为下一代差异化的双靶眼科用药,RC28在DME和DR两个适应症竞争格局良好且研发进度居第一梯队,我们预计RC28将于2024年~2025年在国内获批上市,在考虑上市成功率和降价预期的情况下,2030年销售额预计将达到27亿元。
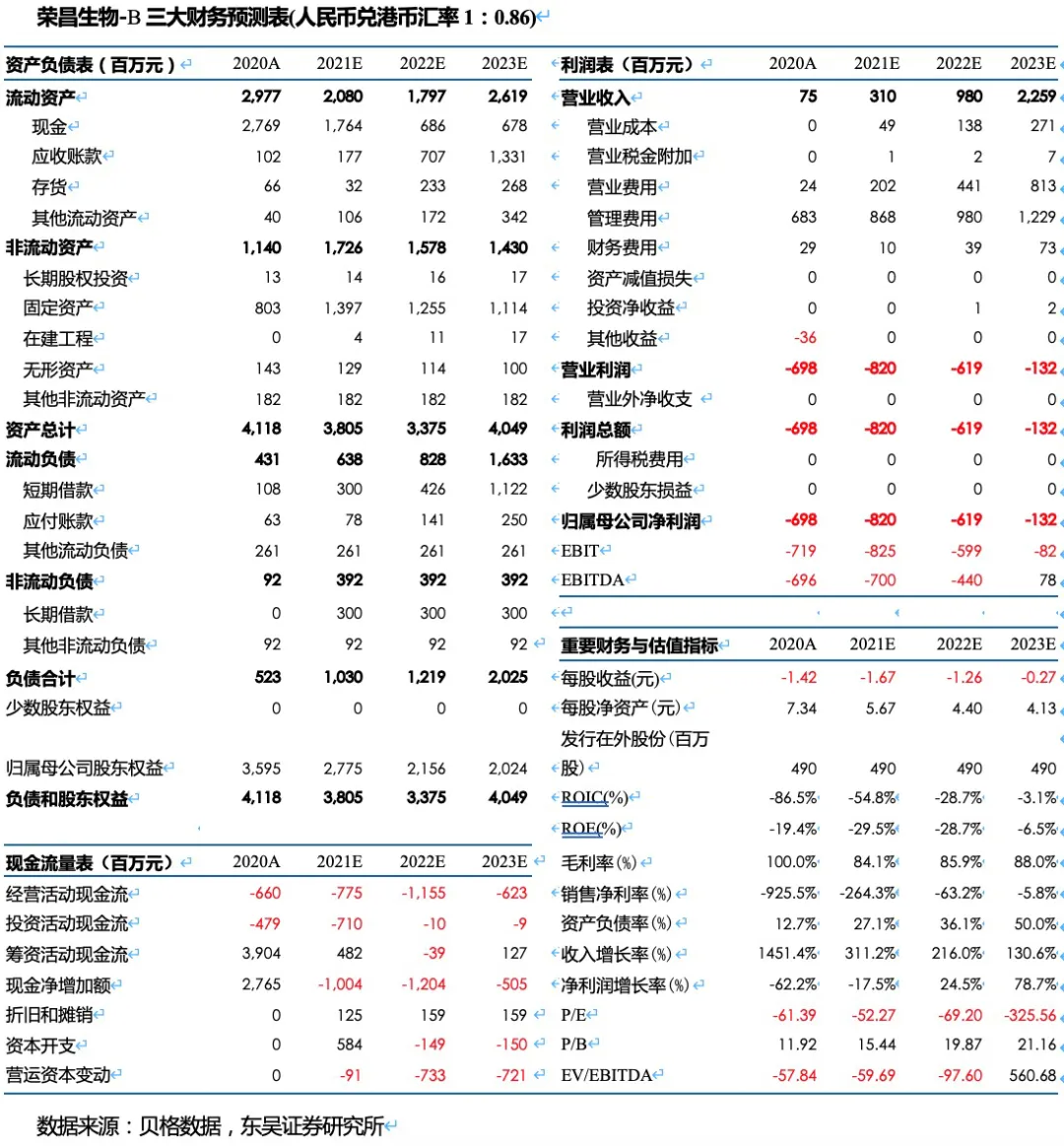
盈利预测与投资评级:我们预计2021~2023年荣昌医药-B(09995)实现收入3.10、9.80、22.59亿,对应归母净利润-8.20、-6.19、-1.32亿。根据测算,我们预计公司将于2024年实现盈利,2025年归母净利达到8.77亿,对应PE 49倍。以PS计算,2021~2025年收入对应的PS分别为138、44、19、17和10倍。按PS估值法,2030年给予10倍PS,折现后对应2021年目标市值738.7亿港币,首次覆盖给予“买入“评级。



1. 荣昌生物致力于开发具有全球创新性的生物创新药
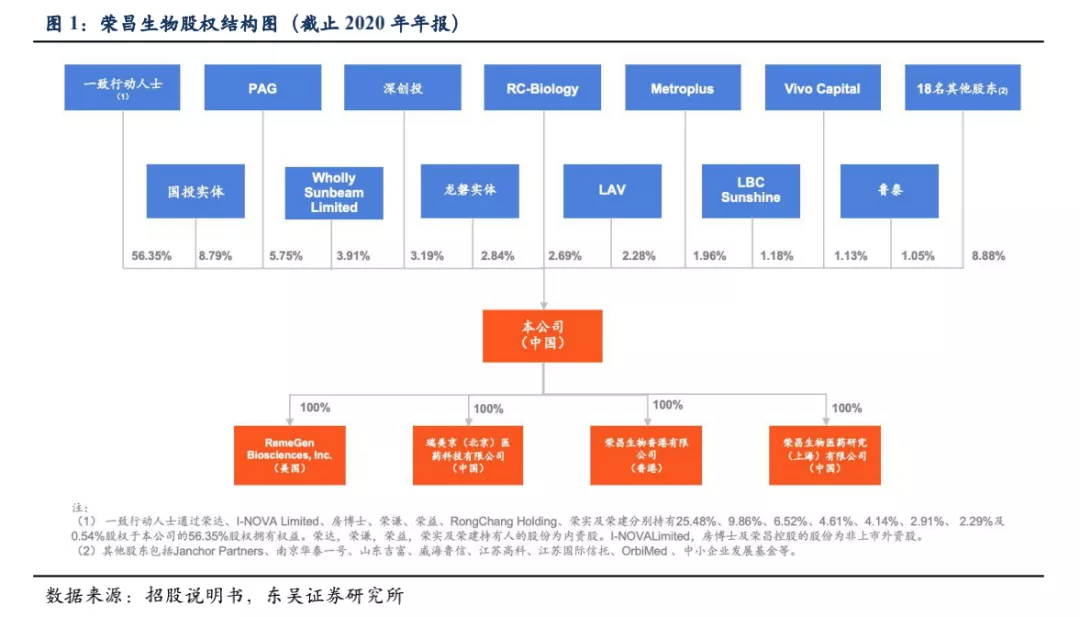
荣昌生物由王威东先生(荣昌制药创始人)和房健民博士于2008年共同创立,成立之初荣昌制药持股65%、房建民博士持股35%。经股改后上市,公司一致行动人及境内外员工股权激励平台持股比例为59.25%(含RC-Biology,荣昌生物境外雇员持股平台)。
1.1 三大领先的研发平台助力公司可持续发展
荣昌生物拥有抗体&融合蛋白、ADC(抗体偶联药物)和双功能抗体三大研发平台,针对自身免疫、肿瘤、眼科等重大疾病领域研发First-in-class和Best-in-class的生物创新药。根据公司年度报告,公司现有产能1.2万升,2021年新产能达产后将增至3.6万升。(1) 抗体及融合蛋白平台:在Fc融合蛋白的生物信息学辅助蛋白设计及工程方面拥有广泛能力,可持续高效地发现以及改良药物结构。涉及药物RC18(泰它西普,风湿免疫疾病用药)和RC28(VEGF/FGF双靶点眼科用药)。(2) ADC平台:涵盖ADC药物开发生产的整个过程包括抗体合成、连接子及有效荷载。通过筛选多种偶联方式、连接子及荷载的组合,优化ADC结构。开发了专有的“Thiel-bridge”偶联技术,以生产更为同质化的ADC药物,从而有利于改善药理作用和治疗窗口。涉及药物RC48(维迪西妥单抗,HER2靶点)、RC88(间皮素靶点)、RC108(c-Met靶点)、RC118(靶点未披露)。(3) 双功能抗体(Hibody)平台:采用独家研发的新型分子形式(已提交发明专利申请)开发双功能抗体,可克服双抗研发过程中可生产性差的问题,从而实现大规模生产同质化的双功能抗体。涉及药物RC138、RC148、RC158,预计今年会有双抗药物申报IND。
1.2 管线产品具有极强创新性,针对未满足的临床需求,市场潜力较大
荣昌生物的研发管线均为具有全球领先性的生物创新药,具体包含三个处于临床晚期的核心品种:△ 抑制B细胞活化及抗体形成的双靶点融合蛋白——RC18(泰它西普),针对多种风湿免疫类疾病,我们预计2030年销售额超过114亿人民币(详见表1的测算);△ Her2抗体偶联药物(ADC药物)——RC48(维迪西妥单抗),针对多种HER2表达异常的实体瘤,我们预计2030年销售额接近24亿人民币(详见表2的测算);△ 抑制眼部血管生成的双靶点融合蛋白——RC28,针对眼科黄斑病变等疾病,我们预计2030年销售额达到27亿人民币(考虑了上市成功率及降价预期,详见表3的测算)。
几个核心产品瞄准患者人数超百万的大适应症及未满足的临床需求。凭借优秀的结构设计,公司产品具备优于竞品的药物性质和治疗效果,在多个临床试验中展现出优异的临床数据。2021年3月9日,泰它西普(RC18)治疗系统性红斑狼疮(SLE)适应症已在国内批准上市,另有6个适应症处于III期临床或注册性II期临床阶段,同时在美国开展SLE的III期和IgA肾炎的II期临床试验。维迪西妥单抗(RC48)治疗HER2过表达胃癌适应症已提交NDA,并被纳入优先审评通道(我们预计2021年6~7月在国内获批上市),同时正在美国开展HER2过表达晚期尿路上皮癌和胃癌的II期临床试验。此外,管线内在研品种间皮素ADC(RC88)和c-Met ADC(RC108)均处于I期临床,我们预计2022年进入II期临床,同时RC118(保密靶点ADC药物)及双抗药物将在今年内申报IND。

2. 泰它西普:面向风湿免疫大赛道,全球化指日可待
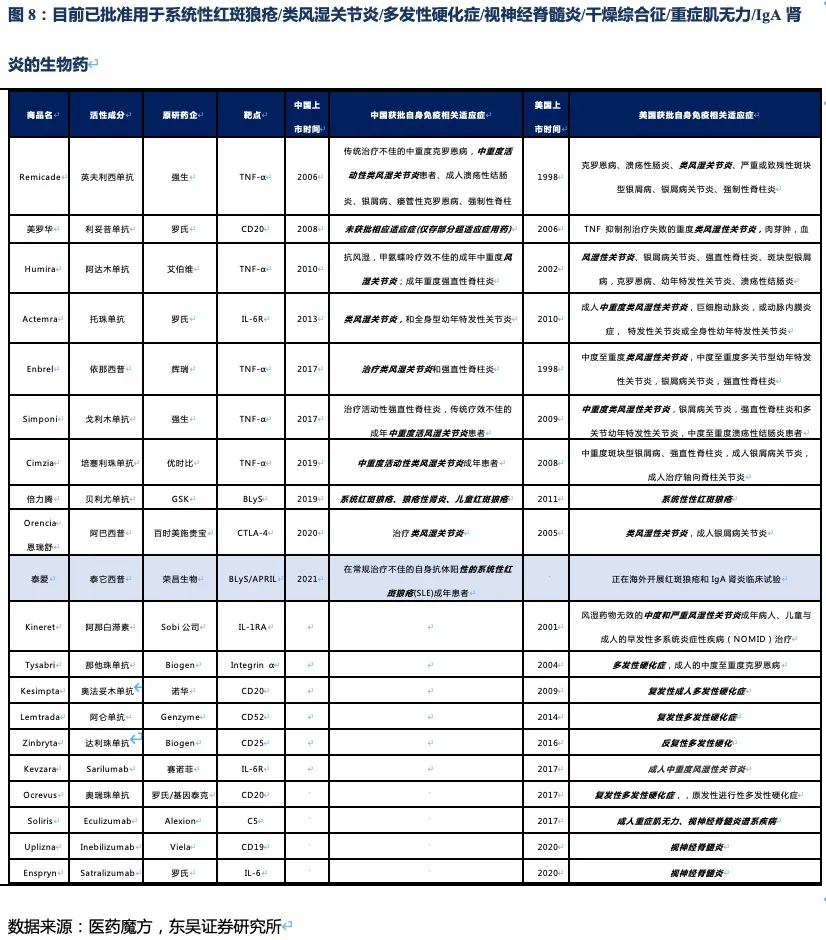
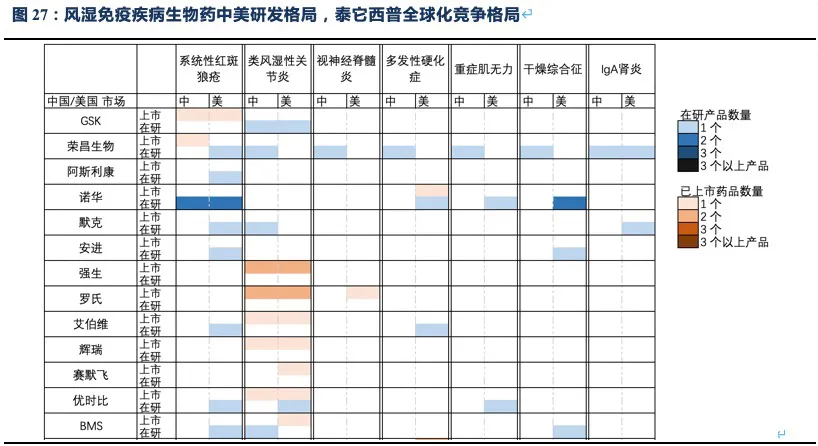
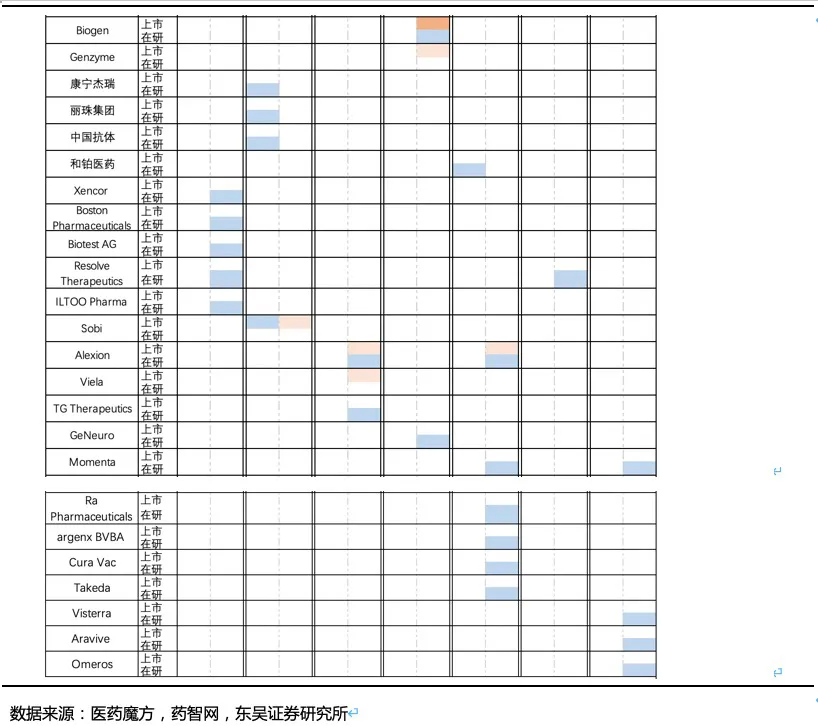
核心观点:泰它西普是全球唯一上市的治疗风湿免疫疾病的双靶点融合蛋白,可以同时靶向2个B细胞发育、增殖分化以及抗体形成过程中非常重要的靶点BLyS(也称为BAFF)和APRIL,从而有效治疗由于B细胞过度活化而引起的风湿免疫疾病。泰它西普已在多个适应症中开展临床试验,在首个上市适应症系统性红斑狼疮(SLE)中,泰它西普相比安慰剂展现出极好的治疗效果(临床应答率:79.2% VS 32%),并于2021年3月9日在国内获批上市,成为全球首个双靶点风湿免疫生物创新药。此外2020年4月泰它西普获得FDA快速通道资格,其SLE适应症在美国直接开展III期临床试验,预计2023年前完成该III期临床。泰它西普所针对的六个核心适应症在国内外的竞争格局良好,尤其在SLE领域仅有GSK的贝利尤单抗唯一生物药竞品,在IgA肾炎和干燥综合征领域的研发进度也居全球领先地位,近5年内都不会出现竞品。鉴于国内风湿免疫市场生物药占比远低于世界平均水平(28.6% VS 68.4%),泰它西普在国内外的百亿销售潜力行则将至,我们预计2030年泰它西普在国内外销售额合计将超过114亿人民币。

2.1 严重威胁健康且无法根治的慢性病魔——风湿免疫疾病
2.1.1. 风湿免疫疾病概述:种类多样,发病机理复杂且无法治愈
风湿免疫疾病是由于人体免疫系统过度活化,无法正常识别外来异物和自身正常组织,而错误攻击自身正常组织导致的疾病。风湿免疫疾病是临床中的疑难病症高地,包括常见的一百多种疾病,其中一类是强制性脊柱炎(AS)、类风湿关节炎(RA)等局部异常炎症免疫反应;另一类是系统性风湿免疫病如系统性红斑狼疮(SLE)、干燥综合征(SS)等全身性炎症。风湿免疫疾病由于发病机制复杂具有“千人千面“的特点,存量患者人数庞大、生存期长、无法被治愈,患者需长期服用免疫抑制剂,临床副作用大,生活质量受严重影响。
风湿免疫疾病触发因素一般为环境刺激以及本身易感的基因型,患者特定部位细胞凋亡速度过快或机体对凋亡细胞碎片清除不及时,导致自身抗原的大量累积。抗原呈递细胞(APC,如树突状细胞)捕获抗原向T细胞呈递,刺激T细胞活化。活化的T细胞辅助B细胞活化并分化形成浆细胞,浆细胞能够分泌大量针对自身抗原的抗体,自身抗体的沉积是风湿免疫疾病患者组织器官损伤、病情加重的直接原因。
2.1.2. 风湿免疫疾病患者人数众多,生物制剂市场空间较大
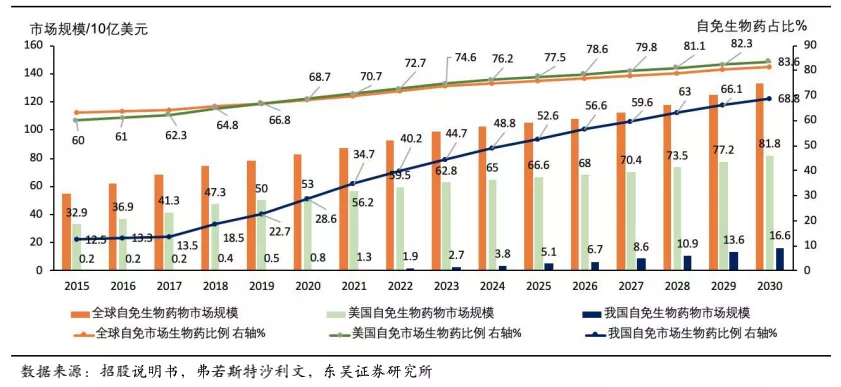
被KEGG 疾病数据库收录的风湿免疫疾病多达100种,影响了全球超过3%的人口。根据弗若斯特沙利文的数据,全球风湿免疫药物市场将由2020年的1205亿美元,增加至2030年的1638亿美元,其中生物药的比例将由68.4%增长至81.4%。美国风湿免疫药物市场规模已在2020年达771亿美元,其中生物药占比也到达了68.7%。而在中国生物药的市场占比远远低于美国仅为28.6%,存在显著的增长空间。随着医保的覆盖、国内患者负担能力的提升,以及医生/患者对生物制剂的认知提升,在中国市场治疗风湿免疫疾病的靶向生物药具有较大的增长潜力。
2.1.3. 风湿免疫疾病治疗现状:对降低激素用量提高生活质量具有迫切需求
风湿免疫疾病无法被完全治愈,只能通过药物缓解症状,防止组织器官进一步损伤。目前针对风湿免疫疾病的治疗手段有限,主要以抑制免疫系统的小分子药物为主,包括糖皮质激素(属于甾体类抗炎药SAIDs)、羟氯喹、甲氨蝶呤、硫唑嘌呤等(均属于改善病情抗风湿药(DMARDs),根据患者病情以及并发症的情况给予合适的剂量及药物组合。传统的非甾体类抗炎药(NSAIDs,如氟比洛芬、萘普生、塞来昔布、美洛昔康等)可以消除关节局部炎症反应,但是只能治标不能治本,不能控制疾病的活动及进展。糖皮质激素起效快、可用于控制急性期病变,重症患者需要长期大剂量服用糖皮质激素,但不良反应多,停药后会复发。常用的DMARDs起效慢、用药数周或数月后症状和体征逐渐减轻,但由于没有明确的靶点,会广泛的抑制免疫系统,因此存在引起感染等风险,感染也成为风湿免疫病患者死亡的重要原因。此外,针对JAK/STAT细胞信号通路的靶向小分子药物JAK抑制剂(如托法替尼、巴瑞替尼等)目前获批的适应症主要是RA,其他在研的适应症包括骨髓纤维化、强直性脊柱炎、银屑病关节炎、红斑狼疮等,还存在较大的不确定性。

2.1.4. 国内风湿免疫学科建设历史较短,是造成生物制剂市场占比低的重要原因之一
我国风湿免疫疾病市场主要被传统的小分子免疫抑制剂等占据,生物药种类少、部分风湿免疫病尚未有针对性的生物药上市,是治疗方案中生物制剂占比低的重要原因之一。另外一个核心原因是我国风湿免疫学科建设的周期较短,缺乏规范的诊疗培训:
我国风湿免疫科是个年轻的学科:风湿免疫学在国际上已有上百年的发展历史,而在我国却是一个新兴的专业学科。1980年我国第一个风湿科建立,2014年启动“一市一科一中心“计划启动(每个县级市至少要有一个风湿免疫专科,每个县级市必须有能够检测风湿免疫疾病抗体及相关指标的中心实验室),2015年,全国只有7000多名风湿科医师,很多医师从其他专科转至风湿科,部分医师还兼顾诊治其他疾病。
近5年内风湿免疫科发展迅速:第四次全国风湿免疫专科医师以及学科调查结果显示,截至2018年9月30日,通过审核的从业人员为12189人,分属于2017家医院的3372个科室,较2015年增长1.6倍,三级以下医院占比由31%增加为45%。其中,有风湿免疫病执业医师的科室较2015年增长1.9倍,专科从业者数量较2015年增长达1.7倍。2019年国家卫健委发布《综合医院风湿免疫科建设与管理指南(试行)》,指出“具备条件的三级综合医院原则上应设立独立的风湿免疫科,鼓励有条件的二级综合医院和其他类别医疗机构设立独立的风湿免疫科“。
医生对生物制剂认知不够充分缺乏规培:1999年,首个生物制剂在国外上市,国内2004年开始将生物制剂用于风湿免疫疾病的治疗。国内医生对传统小分子药物的使用经验丰富,对副作用有成熟的应对策略,部分生物药临床使用时间短,在能够使用熟悉药物控制病情的情况下,医生会避免冒险使用新药物。
患者对生物制剂的接受需要一个过程:很长一段时间里,风湿免疫疾病的治疗药物以激素等小分子为主,很多患者因此摆脱了风湿致残的悲惨命运,但仍有大量患者对小分子药物应答不佳或无法耐受药物的副作用。但生物制剂大多需要注射给药,相比口服的小分子药物,患者长期使用的接受度不够强。生物制剂价格远高于传统小分子药物,患者支付意愿较低。经过2019~2020年RA治疗用生物制剂大幅降价,部分风湿免疫疾病“少药“的局面得到显著改善。
2.1.5. 局部性炎症免疫疾病生物制剂使用较多,而针对B细胞靶点的系统性免疫病缺乏有效的生物制剂
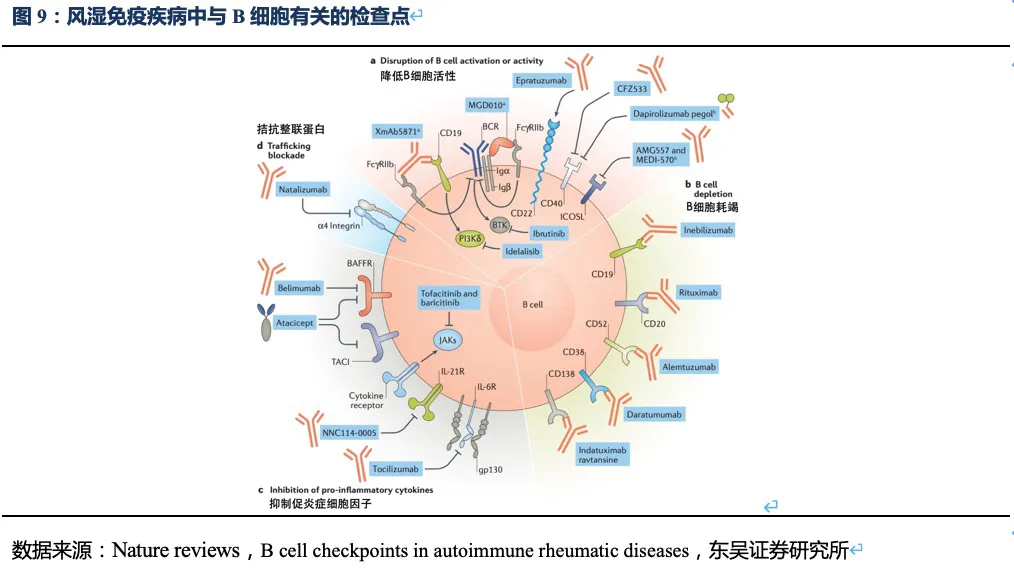
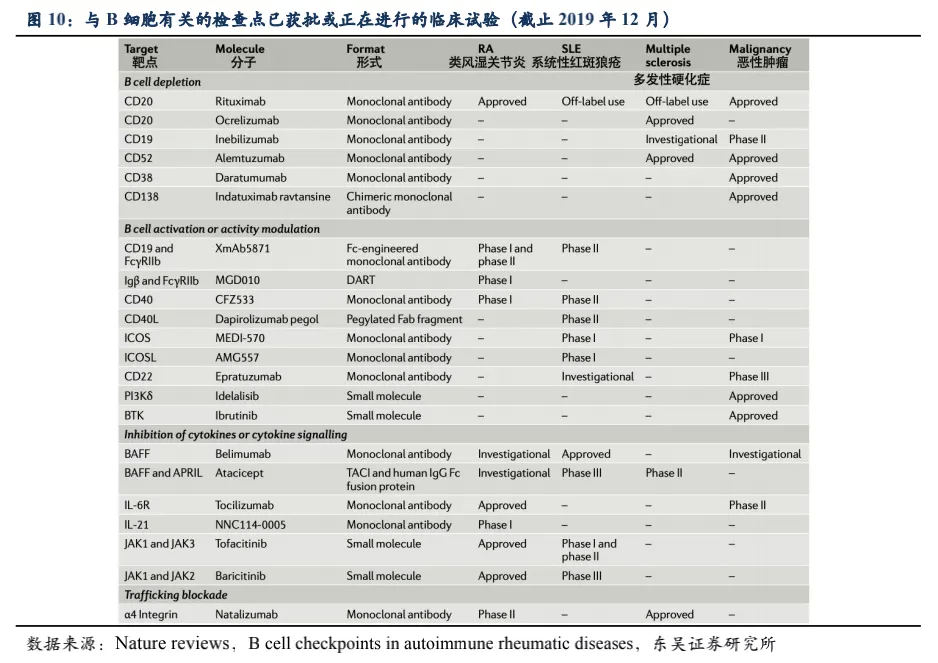
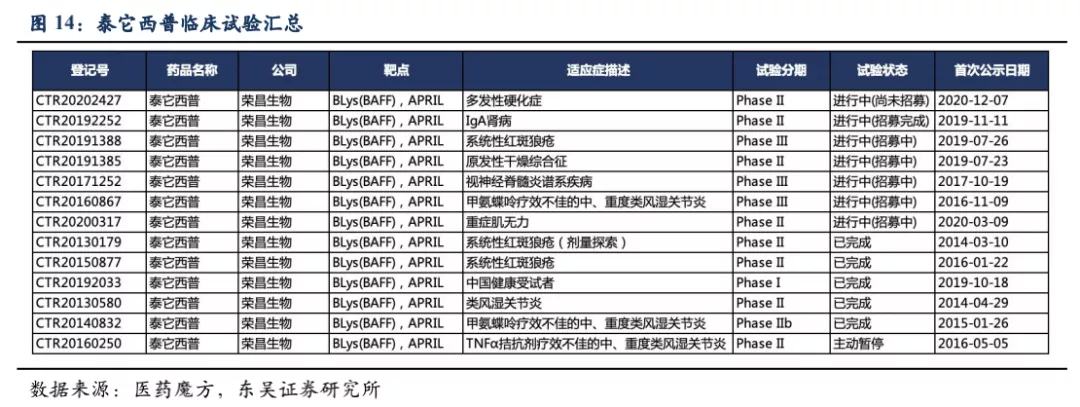
目前的生物制剂主要分为针对炎症细胞因子和直/间接靶向细胞表面受体的两类药物,前者如针对TNF-α、IL-17、IL-23等细胞因子的抗体药物,主要针对局部性炎症免疫反应。常见的直/间接靶向B细胞表面受体用于治疗系统全身性炎症免疫疾病的药物如:① B细胞靶向治疗药物中最著名的是CD20单抗(第一代利妥昔单抗,第二代奥瑞珠单抗),该药物对于RA在国际上有较充分的循证医学证据,但国内没有获批该适应症。利妥昔单抗没有获批MS等适应症,SLE的临床试验也遭遇失败;奥瑞珠单抗2017年被批准用于MS,而SLE和RA适应症因为不良反应停止了临床试验。但该类单抗清除了体内B细胞,有潜在降低免疫力从而造成严重感染的副作用风险。② 2020年在中国上市的RA新药阿巴西普(靶向CTLA-4),从源头抑制免疫活化、阻断T淋巴细胞活化所需的第二信号。与阿达木单抗的头对头研究中显示出非劣效,相比阿达木单抗有更好的应答率,但感染仍是阿巴西普不可忽视的副作用。③ 风湿免疫病新药贝利尤单抗可特异性结合可溶性B细胞刺激因子(BLyS),阻止BLyS与B细胞结合,促进多余B细胞的凋亡,其机理并非完全清除B细胞而是抑制B细胞分化为浆细胞,疗效较为柔和。
根据Nature
reviews杂志针对B细胞表面受体及信号通路的梳理,上市及在研药物中,除了靶向BLyS和APRIL的药物在SLE取得成功外,截止2020年底如以CD19、CD40为靶点的药物在风湿免疫领域进展并不顺利,而JAK抑制剂如托法替尼和巴瑞克替尼针对SLE的临床试验正在进行中,尚未看到其他靶点在短期内可能在系统性风湿免疫疾病取得成功。
2.2. 锋芒毕露的全球首个双靶点B细胞免疫抑制剂——泰它西普
2.2.1. 泰它西普的靶点选择策略和药物结构优势
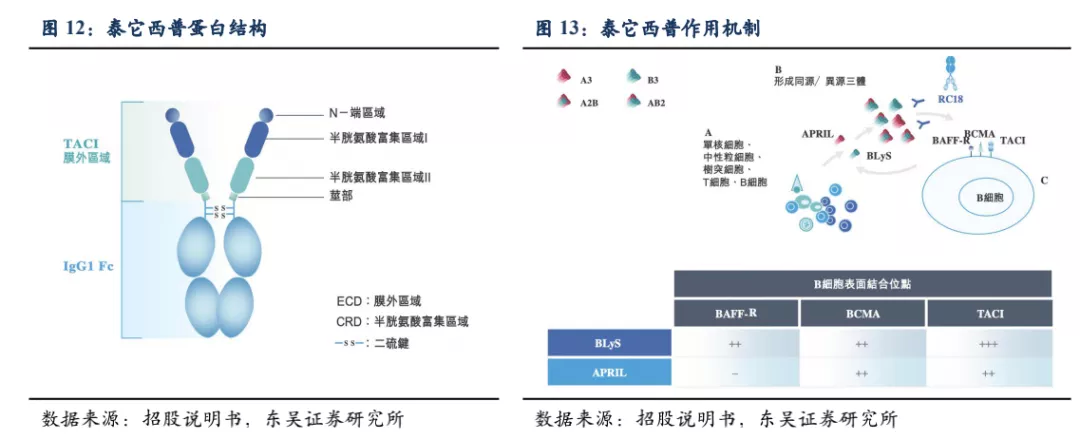
BLyS(也称BAFF)和APRIL是两种跨膜型蛋白、分布在多种免疫细胞表面,是介导自身抗体产生以及自身免疫反应的重要分子,被蛋白水解酶切断成为可溶游离型,通过结合B细胞表面的受体:BAFF-R、BCMA、TACI,刺激B细胞的成熟分化和浆细胞的形成。在这三个受体中TACI对BLyS和APRIL均具有较强的结合力。基于此,荣昌生物将B细胞表面受体TACI的细胞外区域和抗体的Fc段融合,制备成新型全人源TACI-Fc融合蛋白(即泰它西普)。
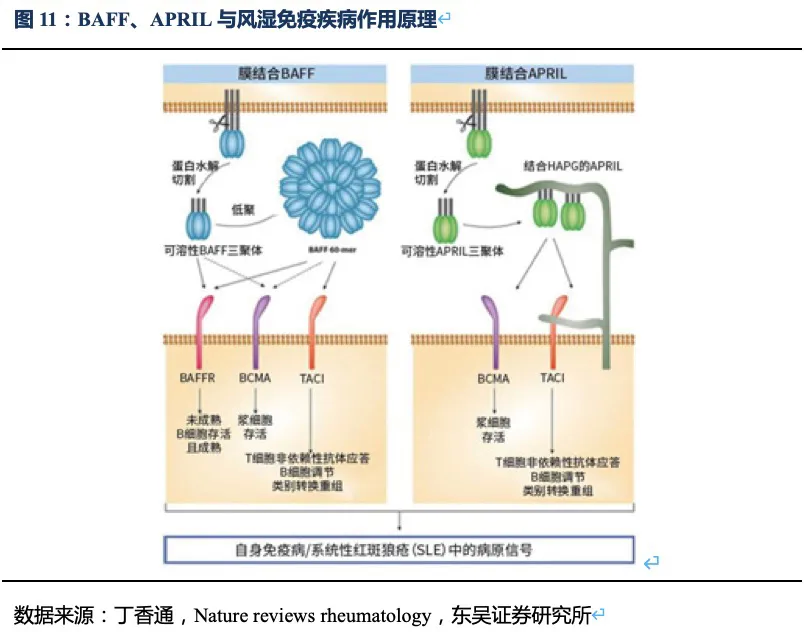
泰它西普的一端是TACI的胞外区域,具有极强的靶向BLyS(BAFF)和APRIL的能力,另一端是IgG1类型抗体的Fc段能够增强融合蛋白的稳定性延长其半衰期。因此泰它西普能够通过双靶机制快速、有效抑制患者体内自身反应性B细胞的发育成熟及自身抗体的分泌。此外,由于泰它西普只会中和三聚体形式的BLyS(BAFF)和APRIL,对于已经与受体结合或者没有形成多聚体的游离单体,并不会产生中和效应,因此只会促进多余B细胞的凋亡,而非完全清除B细胞,并不会产生严重感染的不良反应。
2.2.2. 泰它西普涵盖多个患者基数庞大的风湿免疫适应症,市场前景非常广阔
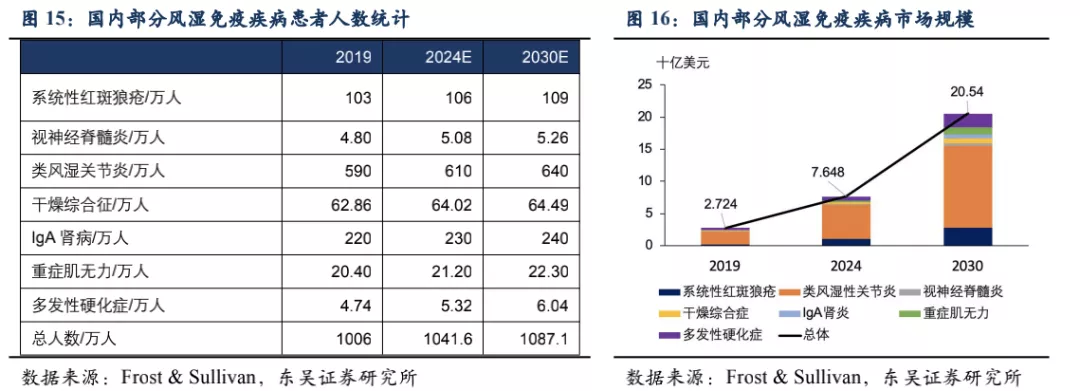
泰它西普对B细胞引起的多种风湿免疫疾病具有潜在的治疗效果,目前已在系统性红斑狼疮(SLE)、IgA肾炎、多发性硬化症(MS)、多发性干燥综合征(SS)、视神经脊髓炎(NMOSD)、类风湿性关节炎(RA)、重症肌无力(MG)等多个风湿免疫疾病中开展临床试验,均已进入II/III期。其中SLE已完成II期临床,于2021年3月在国内正式获批上市。根据弗若斯特沙利文的统计,在我国泰它西普所针对的适应症患者总数目2019年达到1000万人,预计将在2030年达1087万人,针对这些适应症的国内生物药市场总规模将在2030年超过20 亿美元。此外泰它西普于2019年获得FDA优先审评资格,已在美国开展针对SLE的III期临床,以及IgA肾炎的II期临床。根据弗若斯特沙利文的统计,美国SLE患者人数达29万人,尽管患者数少于国内,但美国的SLE治疗市场规模是国内的3倍以上。
2.3. 泰它西普将成为系统性红斑狼疮的最佳治疗药物
2.3.1. 系统性红斑狼疮的疾病特征,及患者对生物制剂的核心诉求
SLE是最典型的系统性风湿免疫疾病,弗若斯特沙利文报告显示,我国患者人数达百万,美国约29万人,且多发生于育龄期女性,Nature review统计女性患者比例高达90%。SLE患者的自身抗原为全身组织广泛分布的核酸和蛋白,因此患者的消化系统、心血管系统、神经系统等多个器官组织均会受到免疫系统的攻击,从而引发一系列复杂多样的临床合并症。为了量化SLE患者的病情,国际上采用SLEDAI-2K标准根据患者的临床表现计算疾病活动程度指数(SLEDAI),用于量化患者疾病严重程度(评分标准详见附录)。SLEDAI<5分的患者疾病基本无活动,SLEDAI处于5-9分的患者为轻度患者,10-14分患者为中度患者,大于15分为重度患者。 SLE患者的一般用药方案:轻度患者推荐长期使用羟氯喹,中重度患者通常采用糖皮质激素治疗,并辅以其他类型免疫抑制剂,常用的包括霉酚酸酯、来氟米特、他克莫司、环孢素以及细胞毒性药物,如环磷酰胺、甲氨蝶呤、巯唑嘌呤等。当以上措施效果不显著,患者反复发作病情无法控制,或者上述药物患者不耐受时推荐使用生物制剂。
临床医生和患者对生物制剂的核心诉求包括:➡
预防病情的反复发作。SLE无法治愈,尽管小分子免疫抑制剂能够延长患者生存期,但患者病情容易反复。协和医院的一项调查显示,SLE患者的年复发率为13%-15.7%,复发也是SLE患者死亡的独立预测因素。《2020年中国系统性红斑狼疮诊疗指南》指出SLE患者治疗的长期目标为预防和减少复发。➡
治疗常规免疫抑制剂无法控制病情的中重度患者。中重度患者需要大剂量使用糖皮质激素或其他免疫抑制剂,部分患者因身体原因无法耐受,或对治疗不敏感,病情无法得到有效控制。➡
减少激素用量,提高生存质量,减少副作用。糖皮质激素的长期使用,会大大增加患者心血管疾病、骨质疏松、内分泌紊乱、代谢疾病的发生风险,对患者身体机能的伤害及外观的影响是显而易见的。2019年欧洲抗风湿病联盟(EULAR)指南指出:SLE治疗要注重症状缓解及维持低疾病活动,同时维持尽可能低的激素用量。➡
育龄期女性患者有生育要求。SLE患者多为育龄期女性,常见的小分子免疫抑制剂常伴有致畸性,必须停用至足够安全的时间,且患者病情稳定至少6个月以上不复发无重要脏器受损,才可考虑妊娠。
综上,SLE患者针对有效减少病情发作,预防脏器受损,减少激素使用剂量,降低激素副作用,对生物制剂具有迫切的需求,特别是有妊娠需求的育龄期女性患者及中重度患者。
2.3.2. 泰它西普SLE适应症治疗效果优异,可以满足患者对生物制剂的核心诉求
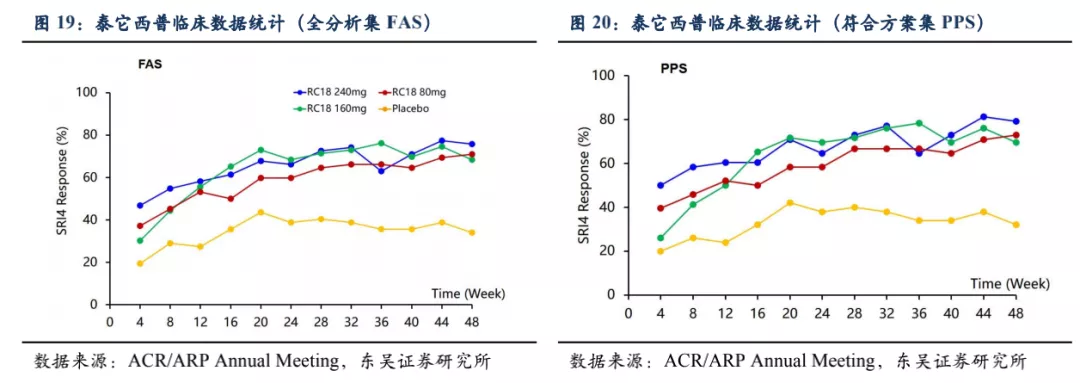
2019年ACR/ARP Annual Meeting泰它西普临床数据公布,249个SLEDAI大于8的中重度患者在接受标准治疗的同时随机接受80mg (n=62)或160mg (n=63)或240mg (n=62) 泰它西普或安慰剂 (n=62) 治疗。在治疗的4~48周之间统计SLEDAI下降4个点的患者的比例(SRI4),以此来量化不同组别的治疗效果。结果显示:
泰它西普对病情的缓解效果好:使用泰它西普的患者比安慰剂组,在相同治疗时间内的响应率显著更高。泰它西普起效速度快:达到40%的响应率泰它西普只需要不到8周的时间,而安慰剂组则需要至少20周。
泰它西普能够控制复发:从已公布的响应率曲线看出安慰剂组在20周后响应率有所下降,表明部分患者病情加重或复发,而泰它西普组患者随着治疗时间的推移响应比例持续升高,说明患者可以产生持续应答。
有潜在的帮助育龄期女性患者妊娠的作用:泰它西普的临床试验结果中发现,11名患者在接受泰它西普治疗后健康状况得到改善,使得她们能够在试验期间怀孕,其中一名患者顺利完成妊娠产下胎儿。
2.3.3. 泰它西普SLE适应症竞争格局:大幅领先上市竞品,在研竞品预计难以超越
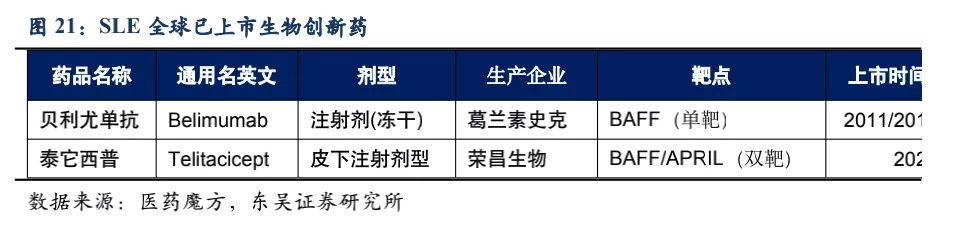
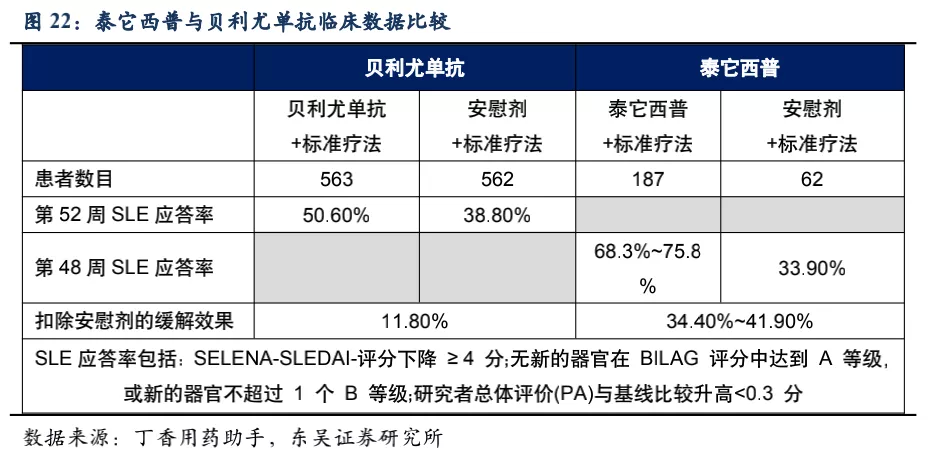
系统性红斑狼疮50多年以来没有新药上市,直到2011年贝利尤单抗在美国上市(2019年10月在中国上市),目前泰它西普的竞品只有贝利尤单抗。从临床数据来看,使用贝利尤单抗第52周患者应答率比安慰剂仅有11.8%的微弱优势,而泰它西普在第48周即有34.4%的相对优势。且贝利尤单抗为冻干注射剂,而泰它西普是皮下注射剂型,方便患者在首次就诊后拿药居家自行给药。
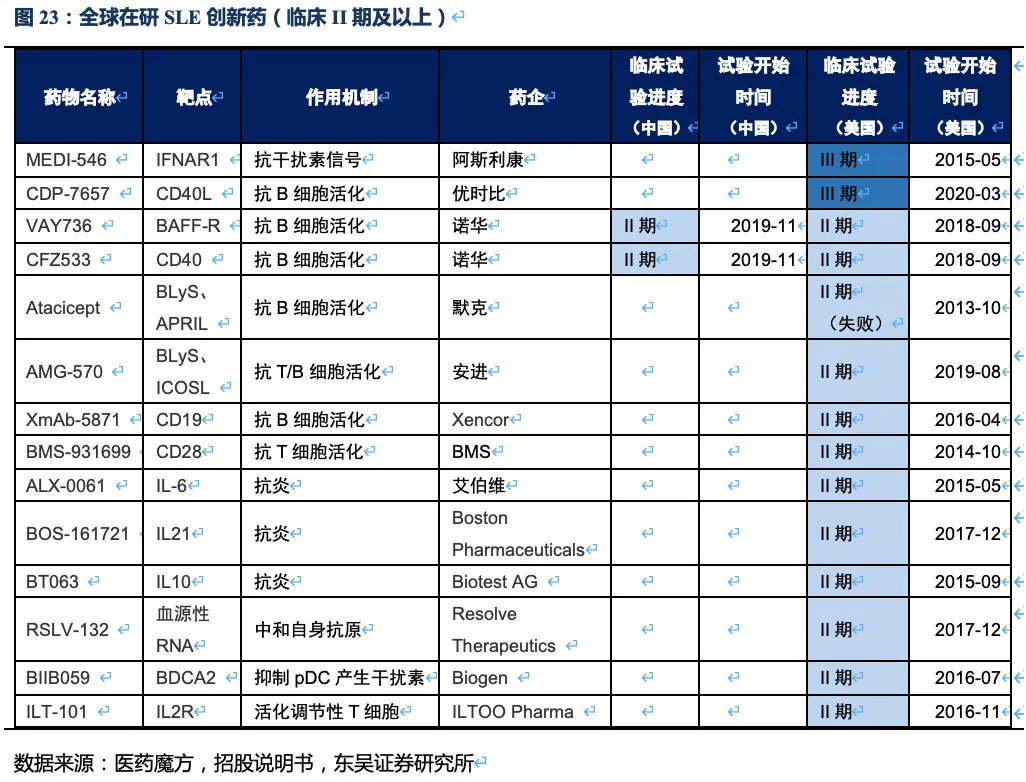
在研竞品大多处于试验前期,进度较快的为阿斯利康的MEDI-546,该药物靶向干扰素受体,用于抑制SLE患者体内过高的干扰素信号。在美国处于临床III期,2020年发表在The
New England Journal of
Medicine上的试验结果显示,治疗52周后患者组响应率为48%(安慰剂组为30.7%),尽管MEDI-546相比安慰剂具有显著的治疗效果,但远未达到泰它西普的治疗效果。综上,泰它西普所面临的全球竞争环境极好,至少5年内已上市及在研竞品均不对其构成威胁。
2.3.4 泰它西普SLE适应症商业化策略:高价高质,目标患者群明确
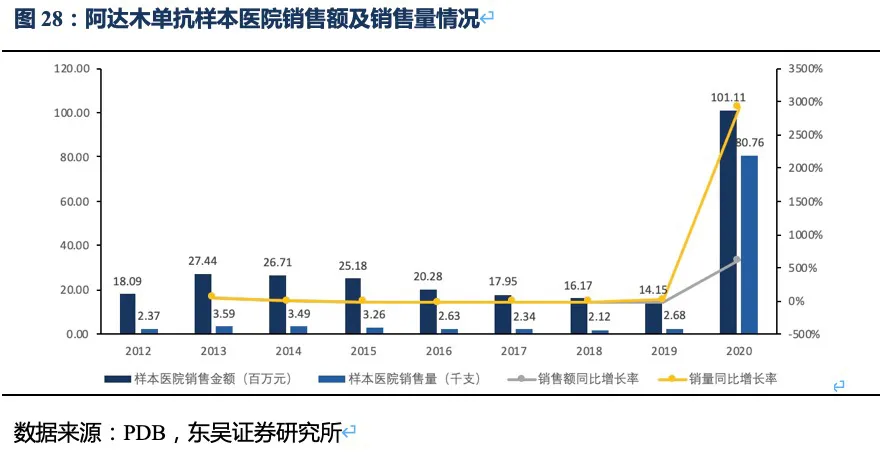
泰它西普上市定价为2586元每支,年用药金额为26.89万元,在患者援助和“爱早享”计划(具体措施见附录)的优惠下,患者年用药金额可降至8.9万以内。2020年贝利尤单抗进入国家医保,单支价格下降62%至755元,年用药金额为5.5万元。我们预计泰它西普2021年进入国家医保后年用药金额将会降至7.10万元左右,表观比贝利尤单抗贵了1.6万元,但由于泰它西普的疗效如应答率指标远好于贝利尤单抗,且部分患者不会足额足量用满12个月,因此并不会比贝利尤单抗造成过高的用药负担。泰它西普目标患者群很明确:(1)中重症或反复发作等传统疗法效果不好,不得不寻求新的治疗方法的患者。(2)本身经济条件好或有妊娠、避免激素使用引起肥胖等外观改变等副反应,寻求降低激素用量提高生活质量的患者。这两大类患者对能够满足其核心诉求的药物,主动或被动的支付意愿最高。
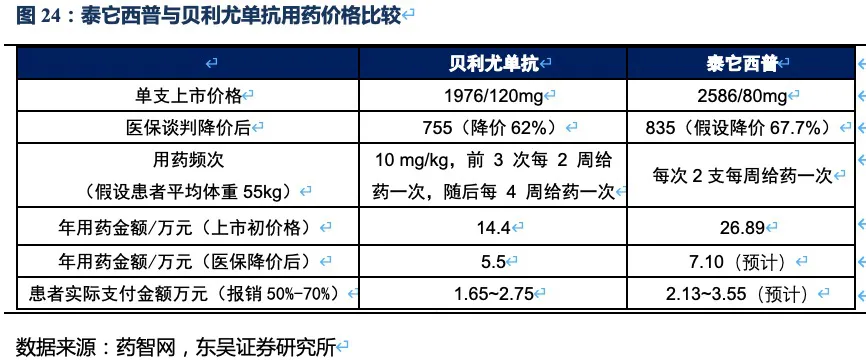
泰它西普销售放量的核心驱动因素包括:目标患者基数大。根据弗若斯特沙利文的资料,我国SLE患者已达103万人,其中适用于泰它西普治疗的患者约37万,并以约1%的速度逐年增加。
能够快速帮助临床医生和患者建立治疗信心。泰它西普治疗效果是已上市产品中最优的,在研品种中至少5年内不会出现疗效更好的竞品。泰它西普见效较快连续使用3个月即可观察到患者的获益(贝利尤单抗需要半年至一年),且大概率不会引起严重感染的副作用,随着治疗效果的快速显现,临床医生和患者对泰它西普的信心增强,有助快速实现放量销售。
重症SLE患者面临重要脏器损伤以及死亡风险,患者对生物制剂的治疗意愿高。我们预计从3月底上市销售起,在没有医保直接报销的情况下,每个月至少有400~500名SLE新患通过援助和“爱早享”计划接受泰它西普的治疗。
不断扩充具有丰富经验的销售团队。目前泰它西普的销售团队约70多人,加上市场人员共100多人,其中超过80%都曾具有自免疾病药物的销售经验,今年底也会根据销售及医保谈判情况将队伍扩充至200人。由于荣昌生物的商业团队在风湿免疫领域有较好的人际资源和市场基础,未来的推广方式也会更灵活多样。
2.4. 其他适应症临床试验推进顺利,竞争局面同样良好
除系统性红斑狼疮外,荣昌也在积极开展视神经脊髓炎(NMOSD)、类风湿关节炎(RA,甲氨蝶呤治疗失败的中重度患者)、IgA肾炎、干燥综合征(SS)、多发性硬化症(MS)、重症肌无力(MG)等系统性风湿免疫病的临床试验,目前均已进入临床II/III期,其中NMOSD和RA已进入临床III期(这些适应症具体发病机理及疾病介绍见附件)。
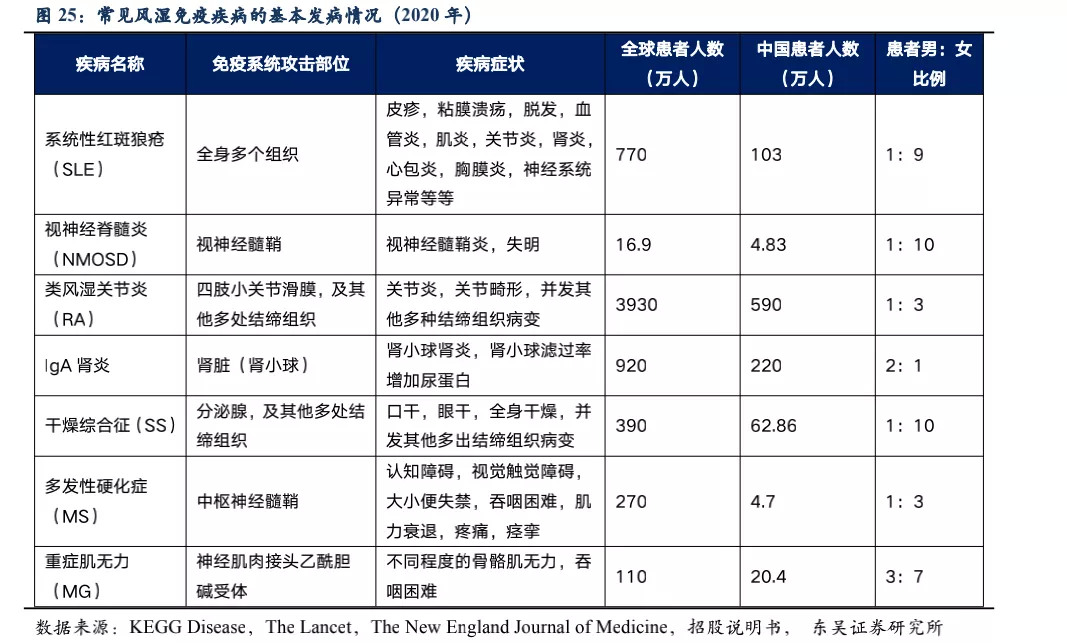
值得注意的是上述疾病中除RA外,我国未批准任何生物创新药上市,国内在研药物种类也少,泰它西普的竞争格局非常优异。在海外市场,虽然在研品种较多,但并不存在同靶点的在研品种,且干燥综合征和IgA肾炎两个较大的市场,绝大部分在研品种均处于临床II期。RA患者人数众多,已批准上市的生物创新药种类也较多,由于阿达木单抗医保降价后患者月自费仅1000元不到,而泰它西普在RA适应症上锚定的人群为甲氨蝶呤治疗失败的中重度RA患者,与阿达木单抗等抗TNF-α生物药不存在直接竞争关系。预计RA适应症也并非泰它西普的核心“战场“,而是通过对RA患者细分探索出一个适用情景,以降低RA市场的推广难度。
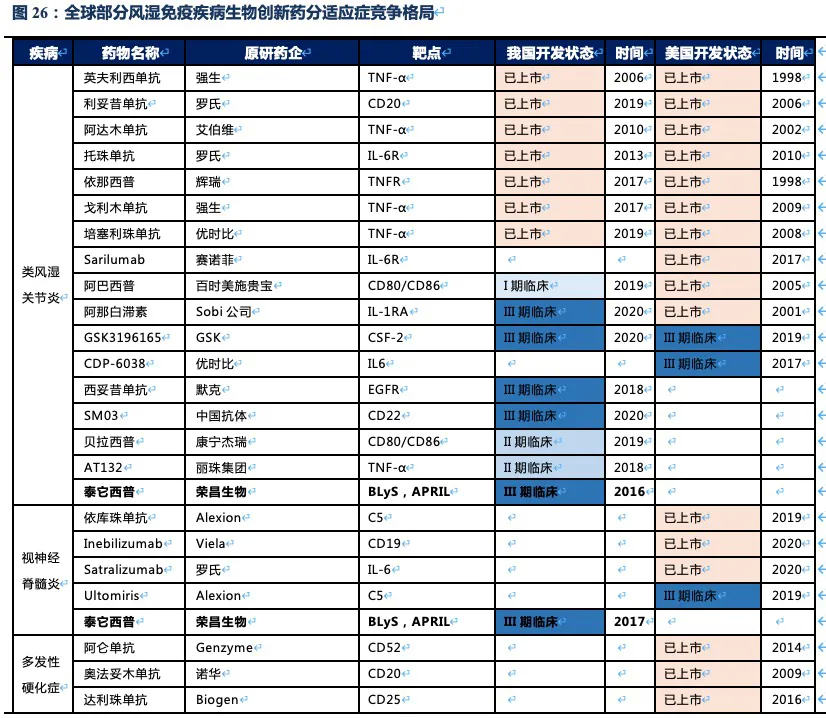
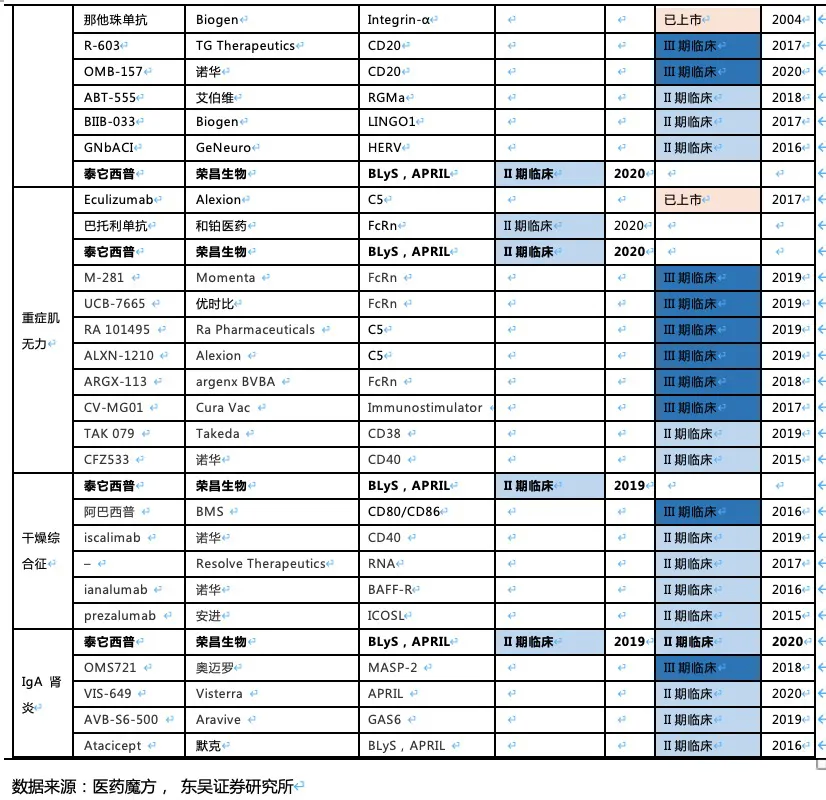
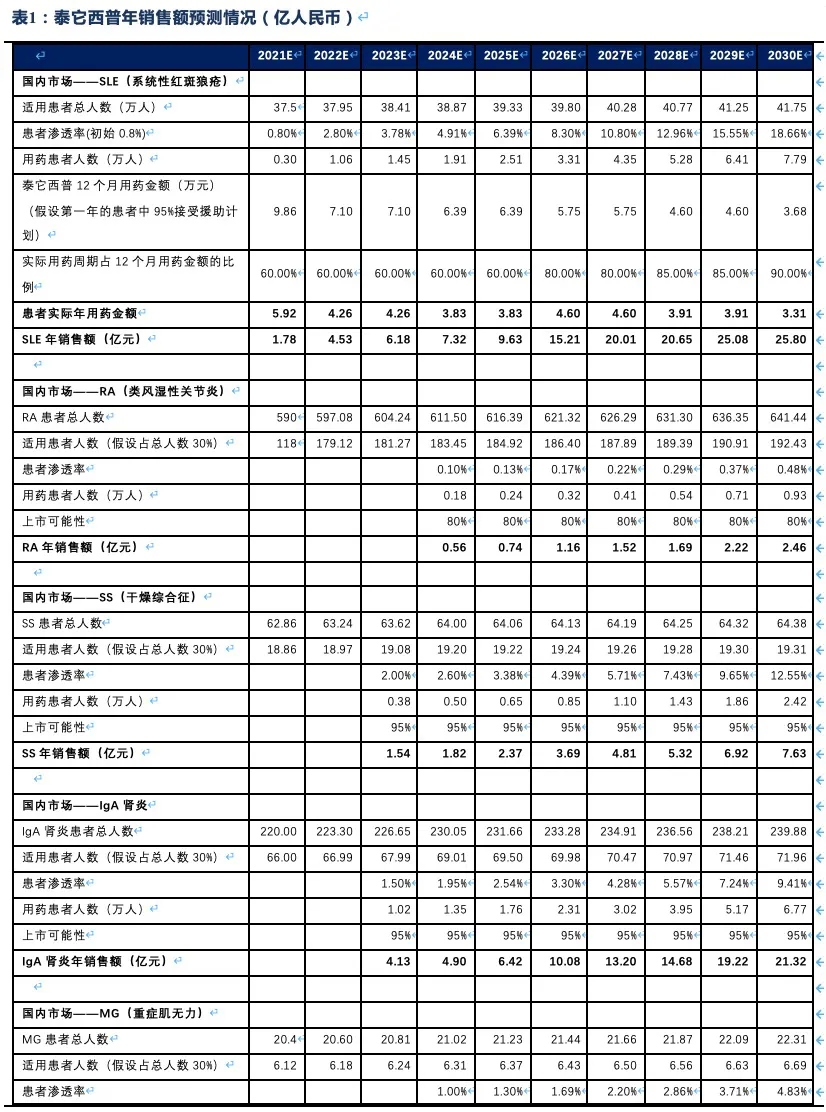
2.5. 泰它西普2030年销售额有望突破114亿人民币
我们对SLE适应症销售额预测假设:
初始销售额测算:根据弗若斯特沙利文的统计数据,我国泰它西普的适用患者约37.5万人,每年以1.2%的速度增加,假设第一年渗透率为0.80%。由于患者援助计划的存在,年用药金额可降低至8.96万元,假设第一年接受治疗的患者95%可享受优惠,5%为原价购买。
未来进入医保报销的影响:2019年底阿达木单抗进入医保后患者年自费金额仅需1万元左右,2020年其销售额增幅达615%、销售量同比增幅达2917%,可见自身免疫疾病市场潜力大,患者对价格较为敏感,当价格降低至一个能接受的水平时,患者治疗意愿显著加强。2021年Q4泰它西普将进行医保谈判,2022年实行医保价格,假设价格降低73.6%,医保后的年用药金额为7.10万元(后续每两年调价一次),以报销70%算,患者仅需自费2.13万元,我们预计泰它西普也将出现显著的销量提升,假设进入医保后对目标患者人群的渗透率增加3.5倍,之后渗透率以每年20%~30%的速度增加。
美国市场测算:泰它西普获得FDA给予的快速审评通道,其SLE
III期临床试验2021年正式启动,假设3年后即2024年获批上市,由于市场售价难以估计,我们以弗若斯特沙利文统计的SLE生物药市场总规模乘以泰它西普的市场占有率计算其在美国的销售额。由于泰它西普所处的竞争环境良好,假设第一年销售市占率可达3%,后续5年市占率以每年30%的速度增加。
我们对其他风湿免疫疾病适应症销售额预测假设:
获批上市时间:SLE适应症从II期临床开始到获批上市一共花了5年时间,目前其他适应症均已进入临床II期,假设从临床试验开始到获批上市需要4-5年时间(部分适应症可能与SLE相似在疗效优异的情况下直接以II期数据有条件获批上市;RA适应症临床试验速度较慢,临床试验于2016年开始,假设2023年上市)。
获批上市的可能性:除了SLE和RA外,泰它西普正在临床试验中的适应症在国内均没有生物药上市,泰它西普较对照药取得“非劣“水平的临床数据即可获批上市,我们将上市可能性的基准定为50%。根据各疾病的致病机理和B细胞的相关程度,以及是否属于系统性风湿免疫疾病,再对不同疾病的上市概率进行调整。
市场渗透率:RA患者中的渗透率和有效性数据、竞争格局以及适用人群大小相关。其他适应症由于尚未有生物药上市,且届时泰它西普也已经在国内商业化推广了2~3年,有助于其他适应症上市初期渗透率的提升,假设这些适应症在适用人群(假设适用人群占总患者的比例为30%)中的初始渗透率均高于SLE的初始渗透率,之后渗透率以每年20%~30%的速度增加。基于上述假设我们对泰它西普未来的销售额进行了测算(2021~2030年):
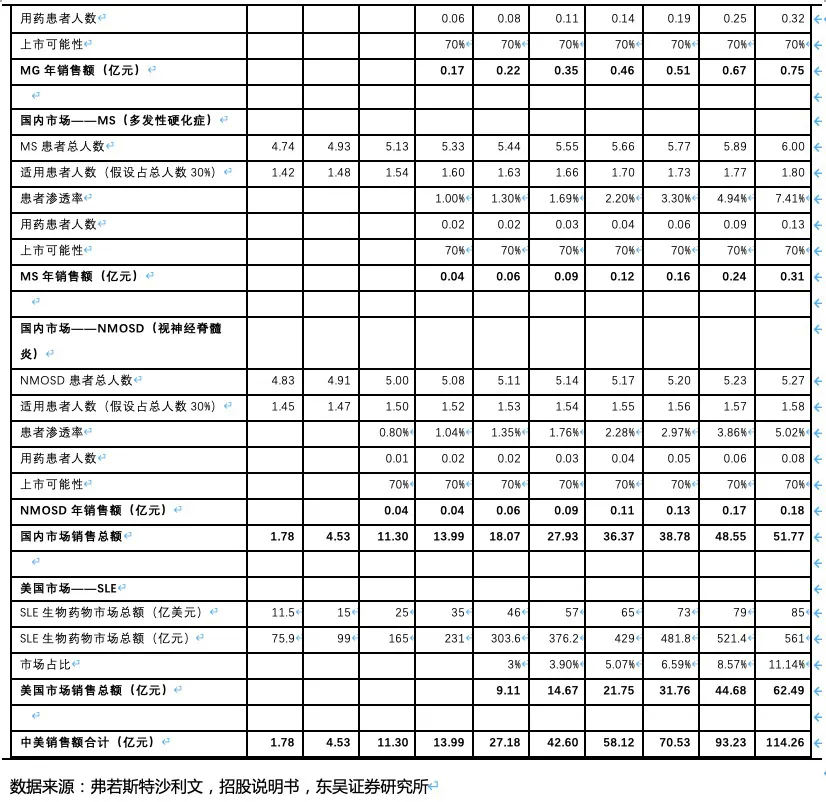
3. RC48:国内最领先的ADC药物研发平台之一
核心观点:抗体偶联药物(ADC)是荣昌生物布局的另一个重点平台。2021年6~7月将在国内上市销售的维迪西妥单抗(RC48-ADC),具有更好的Her2靶向性、更优的血清稳定性、更低的药物毒性、更强的旁杀伤效应。这些优势使得RC48分别在HER2过表达晚期胃癌以及尿路上皮癌的三线和二线治疗中达到24.4%、60.5%的总体缓解率,具有成为最佳后线及二线疗法的潜力。其晚期胃癌适应症已于2020年8月在国内提交NDA,并被纳入优先审评,即将成为国内首个上市的国产ADC药物。此外RC48还在美国FDA获得胃癌临床II期的试验许可和快速通道资格,以及尿路上皮癌突破疗法认定。针对其他多种实体瘤如HER2过表达胆管癌、HER2过表达或突变NSCLC的临床试验也在顺利推进。基于上述若干肿瘤适应症及国内明显的先发优势,仅考虑国内市场,我们预计2030年RC48的销售额将接近24亿人民币。
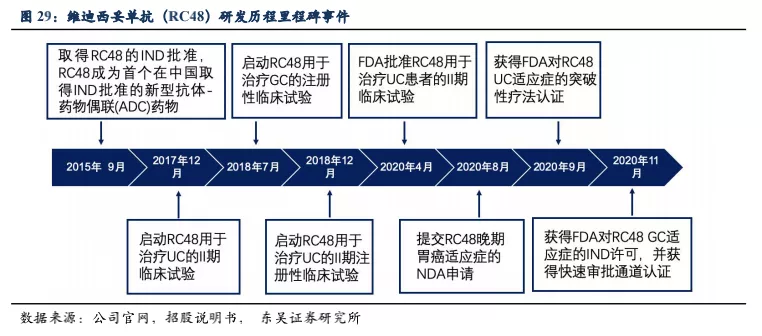
3.1. 高效低毒的肿瘤治疗生物导弹——抗体偶联药物 (ADC)
3.1.1. ADC药物的结构特征,作用机制及临床优势
ADC药物是将肿瘤靶向抗体(Tumor Targeted Antibody)和小分子细胞毒性药物(Cytotoxic Drug)通过连接物(Linker)偶联的一类药物。ADC药物像一个巧妙的生物导弹,抗体部分是定位目标的弹头,细胞毒性药物是弹药,实现对肿瘤的精准打击。ADC药物的作用过程:➡ 靶向:ADC药物的抗体部分和肿瘤细胞表面抗原结合;➡ 内化:细胞发生内吞,将 ADC 药物吞入细胞内,连接物在胞内酶的作用下或溶酶体的酸性环境下分解;➡ 杀伤:ADC上偶联的小分子细胞毒性药物释放,对肿瘤细胞进行毒杀;➡ 旁观者杀伤效应:ADC药物与肿瘤细胞表面受体结合后,借助肿瘤微环境与血液中环境的差异使得Linker断裂,释放的小分子细胞毒性药物具有较好的脂溶性,透过细胞膜弥散在肿瘤微环境中,又会杀死周围原本没有被 ADC结合的肿瘤细胞。 提升药物稳定性和药代动力学以及降低脱靶效应始终是 ADC药物的迭代目标。
ADC药物复杂的结构也导致了其较高的研发难度,一款优秀的ADC药物必须具有以下几个特点:➡ 合适的靶向抗体,具备对抗原(受体)的高度特异性和亲和力,实现对肿瘤细胞精准识别;➡ 结构精巧的Linker,既要具有正常生理环境下的稳定性,又需要在特定环境下裂解高效释放细胞毒性小分子药物;➡ 合适且均一的药物抗体比(DAR),防止不载药的游离抗体大量聚集而被机体清除;➡ 组装好的药物具有高细胞毒性、低免疫原性、低聚合效应、血清稳定性好、偶联能力好的小分子药物。
ADC药物的技术发展历程:一代ADC药物: 使用不可裂解的 Linker和鼠源单抗,抗体靶向性低且具有免疫原性,治疗效果差;二代ADC药物:
使用靶向性更强的抗体,更有效的小分子毒性药物、药效得到提高,但由于不合适且不均一的药物抗体比,导致 ADC 聚集或被机体快速清除,治疗窗口太窄,Linker
问题仍未解决,脱靶效应严重;三代ADC药物:
小分子药物与单抗进行位点特异性偶联,抗体药物比合适且均一性高,拥有很好的血清稳定性和优秀的药代动力学,单抗特异性高,Linker设计更精巧,
脱靶效应和副作用降低。
3.2. 维迪西妥单抗 (RC48) 将成为国内首个获批的国产ADC药物
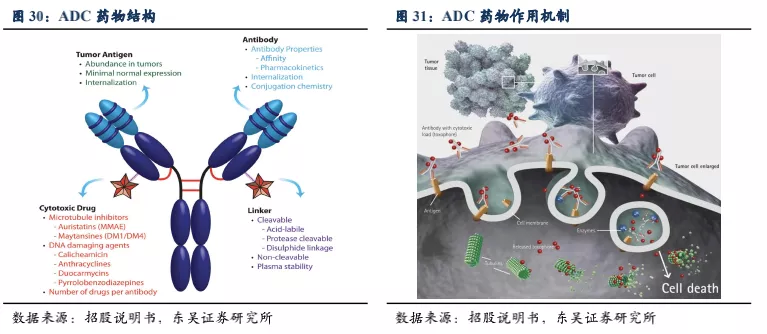
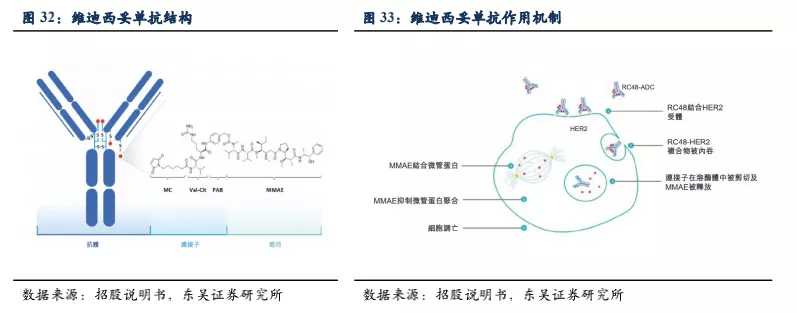
3.2.1. 维迪西妥单抗(RC48)的结构改进和临床优势
ADC药物一直是荣昌的研发重点,韦迪西妥单抗(RC48)是一种新型的人源化HER2抗体和一甲基奥瑞他汀(MMAE)以适当的比例偶联而形成。HER2是表皮生长因子(EGFR)家族的成员,高表达于多种肿瘤组织中,其抗体常用于治疗多种癌症,例如曲妥珠单抗、帕妥珠单抗等。从药物结构看RC48临床优势如下:
靶向性好:韦迪西妥单抗所用的抗体为改良后的HER2单抗,相比曲妥珠单抗,具有不同的抗体结合表位和更高的亲和力及內吞效率,精准导向肿瘤细胞,降低脱靶效应。
肿瘤杀伤效果好:韦迪西妥所偶联的小分子药物为MMAE,这是一种抗微管蛋白聚合的高细胞毒性小分子药物,同时其具有较高的膜通透性,当药物被内吞入细胞释放大量MMAE后,胞内MMAE会更容易从细胞膜透过进入肿瘤微环境中再进入其他肿瘤细胞内,杀伤周边肿瘤细胞。
旁杀伤作用强:韦迪西妥所用的连接子为可裂解的肽连接子,这种连接子会被溶酶体水解酶(这种酶在血浆中的活性极低,因此脱靶效应小,安全性佳)裂解,从而在胞内释放药物。与不可逆连接子比,这种连接子对细胞内吞的依赖低,有利于发挥旁杀伤作用。
3.2.2. 维迪西妥单抗(RC48)的适应症布局
目前,RC48的适应症为实体瘤,包括胃癌、尿路上皮癌、胆道癌、乳腺癌、非小细胞肺癌等,线数为二线及以上的治疗。值得注意的是,即使患者的肿瘤细胞仅仅表达低水平的HER2蛋白,RC48也能够精准靶向肿瘤组织,在肿瘤局部高效释药并发挥旁杀伤效应,因此其临床试验也将HER2低表达的患者纳入其中。由于HER2低表达及阴性患者相比阳性患者,缺少有效的药物,RC48有望成为这部分患者治疗的理想选择。
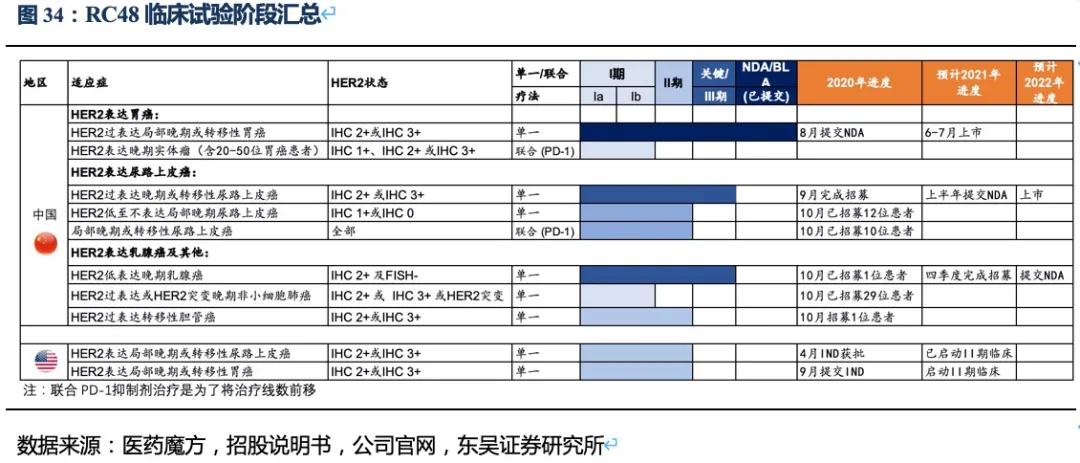
3.2.3. RC48治疗胃癌的竞争格局:国内最佳的晚期胃癌三线及以后治疗药物
胃癌是RC48进度最快的适应症,2020年8月在国内以II期临床数据提交NDA,预计将于今年6~7月获批上市,进入胃癌三线治疗。 胃癌流行病学特征:胃癌是世界范围内最常见的恶性肿瘤之一,Globocan的最新统计数据显示,2020年全球新增胃癌患者109万,东亚是全球胃癌发病率死亡率最高的地区,而我国是胃癌发病大国,年新增胃癌患者近47.9万人(排第三,仅次于肺癌、结直肠癌),5年累计患者人数约68.8万人。根据弗若斯特沙利文的调查数据,其中约22%患者为HER2高表达胃癌。发病率高,意味我国胃癌治疗市场庞大;死亡率高,说明我国胃癌患者缺少有效延长生存期的药物。
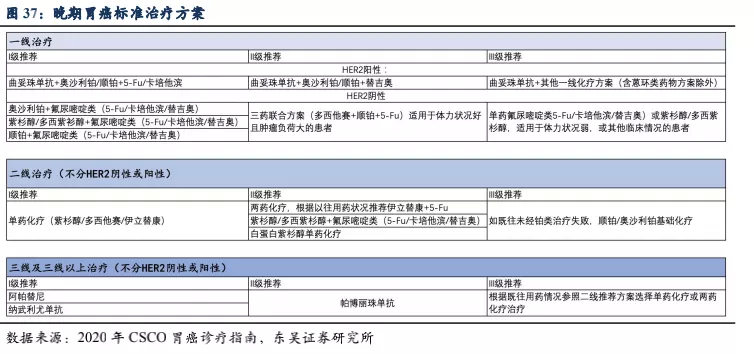
晚期胃癌目前的治疗手段及二线后用药方案:根据CSCO指南,HER2阳性患者的一线治疗以曲妥珠单抗联合化疗药物为主,二线治疗以单药化疗为主,三线治疗主要考虑PD-1单抗和抗血管生成抑制剂甲磺酸阿帕替尼等,可以看出RC48晚期胃癌适应症国内主要竞品为PD-1单抗和化疗药物。
已获批上市的晚期胃癌药物:我国已获批用于胃癌二线后治疗的靶向药包括,甲磺酸阿帕替尼、纳武利尤单抗、帕博利珠单抗。此外美国还批准上市了胃癌二线靶向药雷莫芦单抗(正在我国申请上市),三线治疗ADC药物Trastuzumab
deruxtecan (DS-8201,暂未在我国申请临床,在美国直接推至胃癌的二线治疗)。
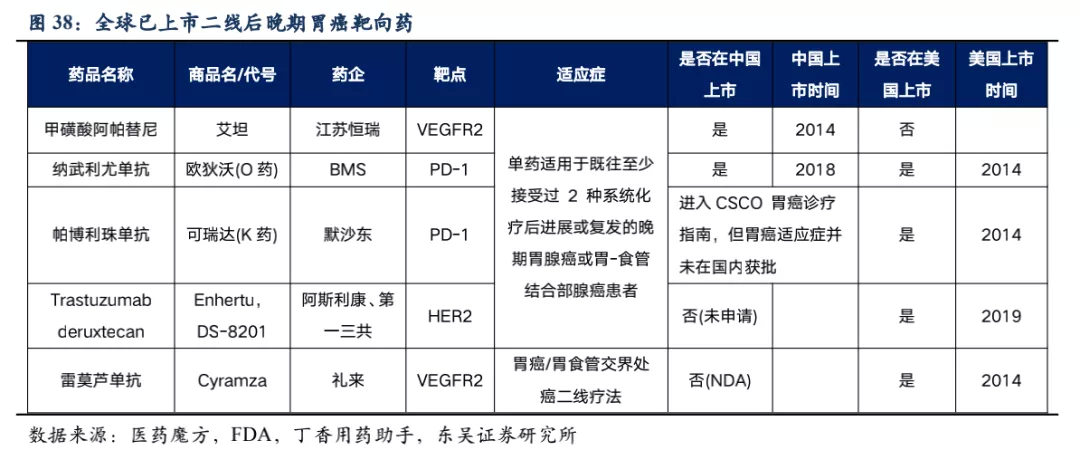
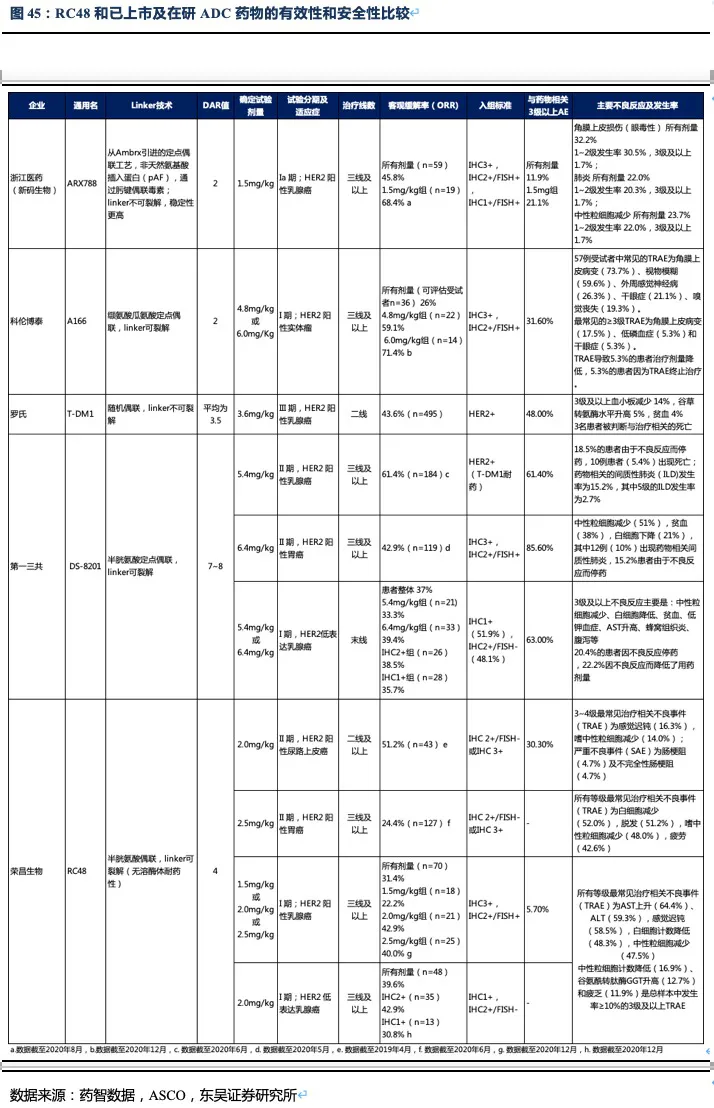
RC48有效性数据领先其他三线治疗药物,有升至二线治疗的潜力,公司积极探索药物联用策略助力治疗线数前移。统计目前二线及以后靶向药物的临床数据,结果显示在二线后药物中,目前在我国上市的阿帕替尼,以及O药单药使用ORR分别不足3%和12%,而RC48的ORR达到了24.4%,此外RC48的中位无进展生存期以及总生存期也同样全面领先国内上市药物。在美国上市的ADC药物Trastuzumab deruxtecan(DS-8201)获批进入胃癌三线治疗(由于疗效超过过往二线标准治疗而直接推至二线治疗),表现出极好的治疗效果,ORR高达42.9%,患者总生存期达12.5个月。
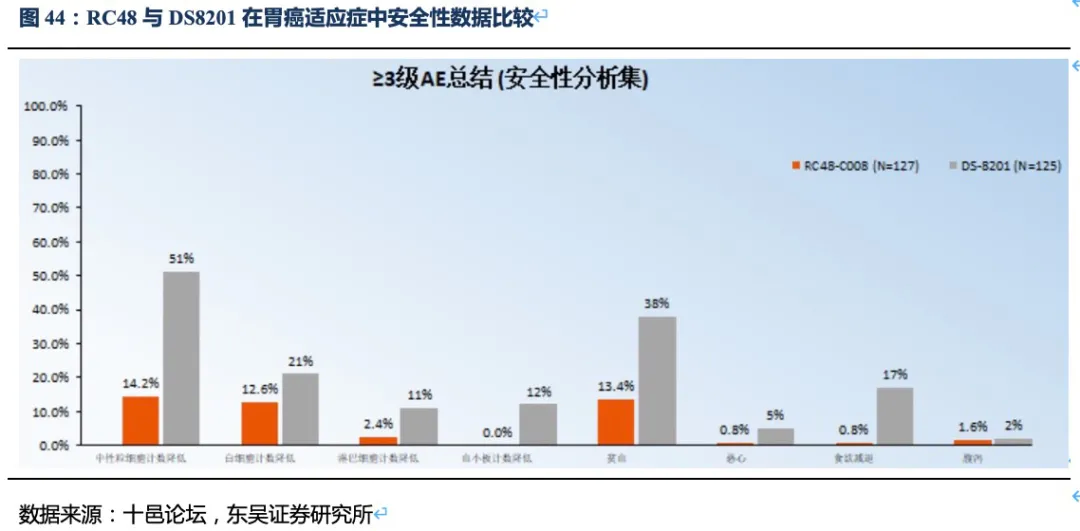
但DS-8201的药物抗体比达到了8:1,如此高的载药率,带来了更好的治疗效果同时其安全性也受到影响。DS-8201在II期临床试验DESTINY-Gastric01中有高达15%的患者永久停药,其中11%的患者发生严重的间质性肺病(ILD),此外在其针对乳腺癌的临床试验中2.6%的患者发生严重ILD,且全部死亡。ILD是一种严重的致命副作用,FDA也因此对DS-8201进行了黑框警告,安全性将会是限制其使用的重要因素。另一方面,DS-8201暂未在我国开展临床试验,RC48上市后在国内竞争环境良好,将成为国内胃癌三线治疗的最佳药物。
根据ASCO等会议结果,目前全球获批用于胃癌二线治疗的靶向药只有雷莫芦单抗,其单药使用安全性良好,和紫杉醇联用ORR为28%,中位PFS、OS分别为4.4个月、9.6个月。这和RC48三线单药治疗的数据相差不大(非头对头),且RC48的II期临床中纳入了部分IHC
2+/FISH-的患者(降低了对HER2水平的要求,扩大了目标人群),说明RC48已经接近胃癌二线标准疗法或化疗的有效性。是否能从同类ADC药物中脱颖而出以及是否可以实现治疗线数的前移,取决于RC48的安全性数据。从已有临床数据看,RC48也展现出良好的安全性(无间质性肺炎或眼毒性产生),治疗线数前移的潜力很大,荣昌生物也在积极探索药物联用(如PD-1单抗)助其线数前移。
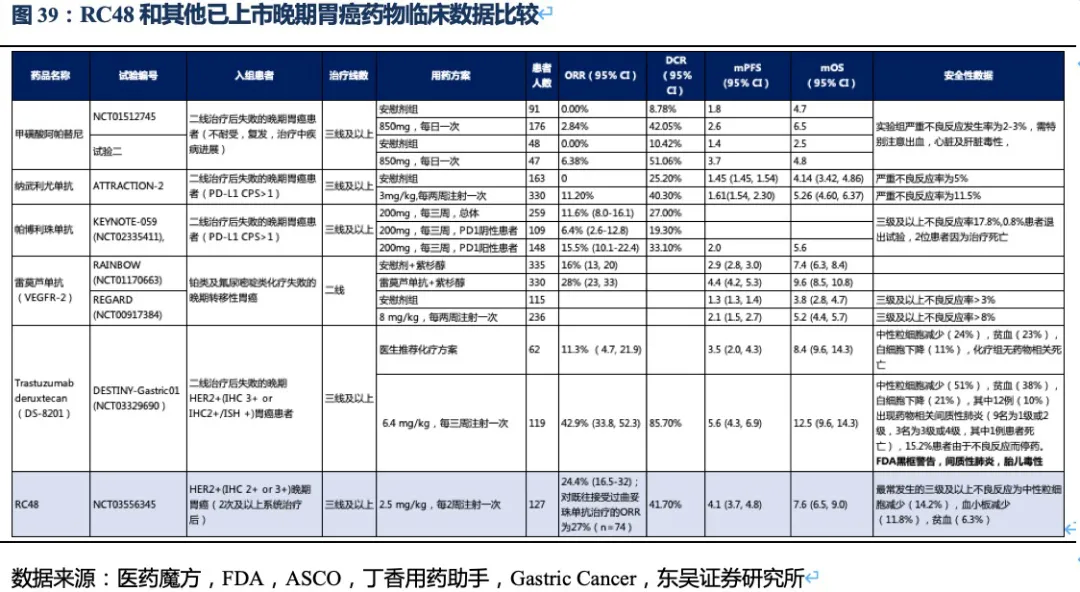
市场认为RC48在胃癌适应症的数据较DS-8201差很多,实际上无论ORR还是OS数据的差异更多是入组患者特征的差异较大导致的,一方面来自人种的差异,另一方面来自患者基线水平和肿瘤特征的差异。而且DS-8201对于HER2低表达胃癌的亚组数据分析结果也并非优于RC48。我们认为RC48完全有能力与DS-8201在部分适应症中竞技,而市场由于简单的表观数据对比低估了RC48的潜力。
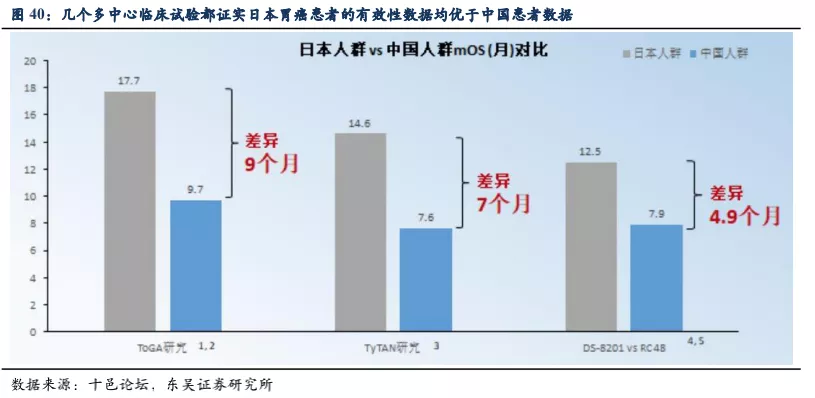
首先,DS-8201的DESTINY-Gastric01试验中入组的患者为日本和韩国人(以日本为主),而从一些历史临床试验比较看,日本患者的胃癌治疗数据都好于中国患者的数据。如赫赛汀联合化疗一线治疗HER2阳性胃癌的ToGA试验(超过一半为日本患者),或拉帕替尼联合紫杉醇二线治疗HER2阳性胃癌的TyTAN试验(超过60%为日本患者),亚组分析中日本患者的mOS都长于中国患者的数据。
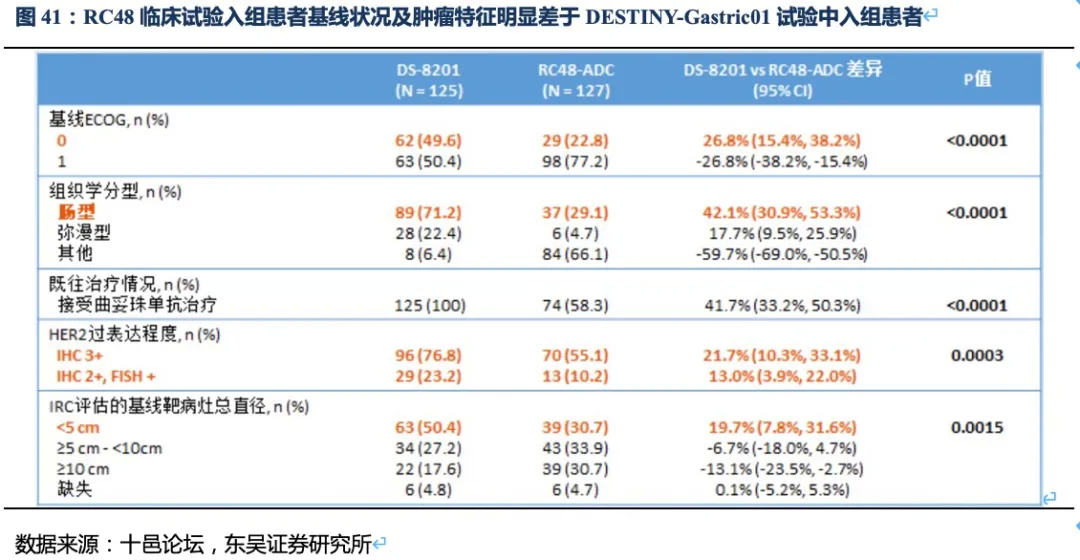
其次,RC48的临床试验入组患者基线状况及肿瘤特征明显差于DESTINY-Gastric01试验中入组的患者。如DESTINY-Gastric01试验中ECOG评分0(评分越低患者的身体状况越好)的患者占比更高;肠型胃癌(恶性程度低、预后较好)占比更高;IHC3+或IHC2+/FISH+占比更多(RC48的临床试验中还有34.7%的患者为IHC2+/FISH-的HER2低表达患者);肿瘤直径<5cm的患者占比更大。因此这些基线和肿瘤特征都会使得DS-8201的临床数据表现更好。
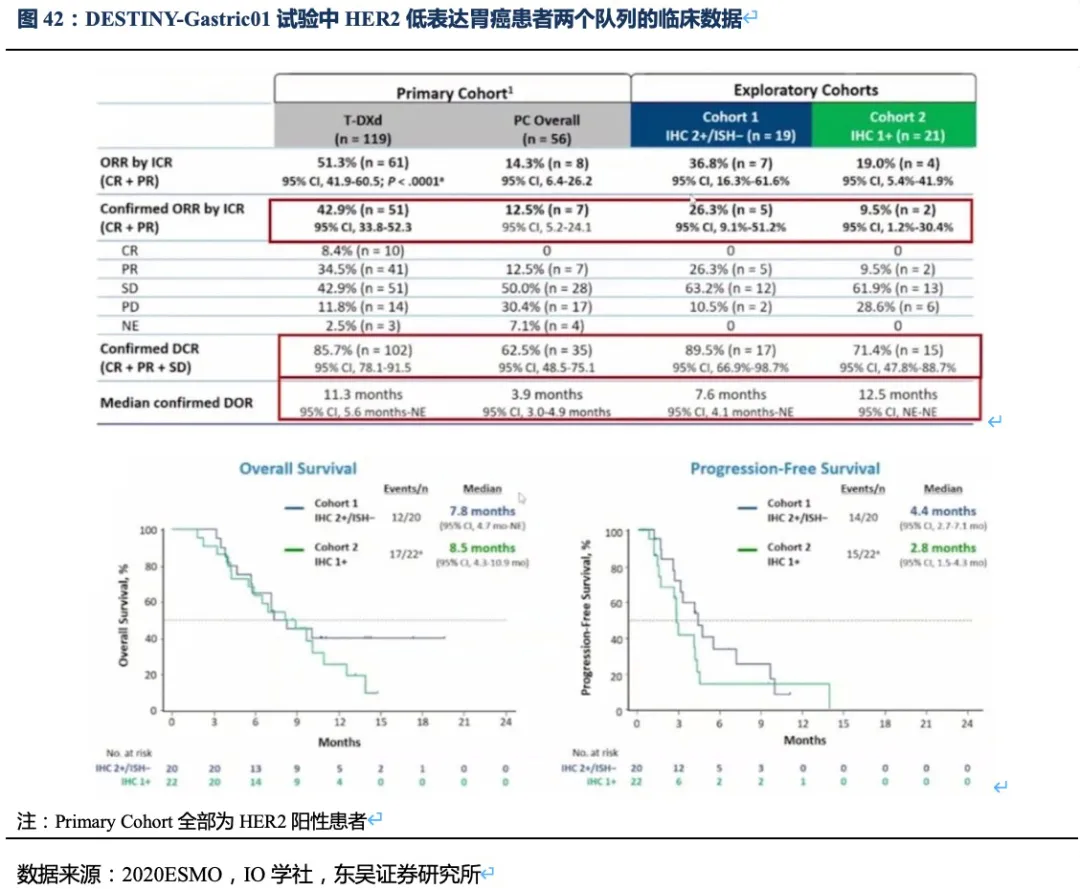
最后,从DESTINY-Gastric01试验的探索队列的数据看,DS-8201对HER2低表达患者的有效性数据并非优于RC48。DESTINY-Gastric01试验探索队列分两个亚组(既往均未接受过抗HER2治疗):队列1(Cohort1,n=20)为IHC2+/FISH-,队列2(Cohort2,n=20)为IHC1+。队列1、2的ORR分别为26.3%和9.5%,mOS分别为7.8和8.5个月,mPFS分别为4.4和2.8个月。
RC48胃癌适应症的在研竞品:针对晚期胃癌的在研药物种类繁多,包括小分子药物、同类ADC、PD1单抗、双抗等产品,目前多个国产PD-1抗体在标准化疗失败后的晚期胃癌中开展临床试验,ORR均在10%~20%之间(根据CSCO诊疗指南统计)。双抗产品(Zanidatamab ZW25 HER2双抗)截止2021年1月只有I期临床数据。ZW25单药二线治疗的ORR为33%(33例患者IHC3+或IHC2+&FISH+),DCR为61%,mPFS为3.6个月,虽然该数据略好于RC48或雷莫芦单抗,但该临床试验样本量较小且针对的是HER2双靶点。百济神州从Zymeworks引进了ZW25于2021年初在国内开展ZW25联合或不联合替雷利珠单抗一线治疗HER2+胆道癌和胃/胃食管链接处腺癌(GEA)的II期临床试验。Zymeworks也于2019年初在海外开展ZW25联合化疗一线治疗HER2+ GEA的II期临床试验。
RC48的主要竞品还将会是其他HER2-ADC药物,国内在研的以胃癌为适应症的HER2-ADC药物,大多于2020年开始进入临床I期,进度较慢。其中最快的为恩美曲妥珠单抗——T-DM1(已在我国获批上市用于乳腺癌治疗),但2016年发表于Gastrointestinal
Cancers
Symposium的一篇摘要指出,T-DM1在二线治疗HER2阳性晚期胃癌患者(n=228)中位PFS仅为2.7个月(对照组为2.9个月),中位总生存期为7.9个月(对照组为8.6个月),和使用紫杉醇的对照组相比,并未发现显著优势。T-DM1组三级及以上不良反应发生率为59.8%(优于紫杉醇组的70.3%)。据此我们认为T-DM1在有效性和安全性方面都难以和RC48媲美。
ADC药物很讲究药物的有效性和毒性的平衡,通过小分子药物的毒性/脂水溶性、Linker的稳定性来实现这种平衡,并不是小分子毒性越高、DAR值越高疗效越好,有时反而会牺牲药物的安全性,导致治疗线数无法前移。从单药研发角度讲,ADC药物的ORR率等有效性很重要,但是从临床疾病治疗的角度,提升ADC药物联用的可行性才是关键,如抗HER2联合免疫治疗是目前探索的重要方向,也因此才会有双抗ADC升级药物的出现。未来,无论胃癌或乳腺癌等与HER2表达相关的肿瘤中,HER2-ADC药物将会成为一种基石用药,其安全性是非常关键的因素。
已有临床数据看,DS-8201的疗效优异,但其出现大比例的严重不良反应,尤其是间质性肺炎的产生,FDA也给予其黑框警告。同样,浙江医药(新码生物)的ARX788和科伦博泰的A166的初步临床数据也显示了较好的有效性(主要是HER2阳性乳腺癌)。ARX788相比T-DM1虽然在肝脏毒性和血液毒性方面降低了,但是出现了肺毒性和眼毒性;而A166也出现了明显的眼毒性。因此,毒性的限制使得上述三种ADC药物治疗线数前移的可能性降低,也就无法形成差异化的竞争优势(如与PD-1联合提升治疗线数)。相比之下,RC48展现出良好的安全性(无间质性肺炎或眼毒性产生),治疗线数前移的潜力很大。目前RC48也在开展与PD-1联用在HER2表达(含低表达)的胃癌和尿路上皮癌的I期或II期临床试验,后续有望实现在胃癌二线、尿路上皮癌一线疗法的突破。
除了有效性和安全性外,我们还需关注到目标人群的扩展。即ADC药物Linker可裂解在异质性较强的肿瘤中,对于HER2低表达或HER2阴性的癌细胞同样有杀伤作用,这是针对这类异质性较强的肿瘤最佳的杀伤策略之一。从几种ADC药物入组患者的特征看,只有DS-8201和RC48入组了部分IHC2+/FISH-或IHC1+的患者(即HER2低表达患者),这样就使得这两种药物不仅可以治疗HER2阳性实体瘤,对于HER2低表达患者也有疗效,目标患者数大幅提高(如在乳腺癌和胃癌中目标患者群扩大了近两倍)。从I期的初步数据看,RC48针对HER2低表达乳腺癌患者的ORR也达到了39.6%,证实RC48对HER2低表达患者同样有效。
结论:我国为胃癌发病大国,晚期胃癌患者缺少有效治疗手段,胃癌的二三线治疗竞争格局良好,RC48将会成为胃癌三线的最佳治疗药物,同时有望通过联合疗法升至二线用药。➡
RC48和中美已上市的其他晚期胃癌三线用药相比,具有治疗效果或安全性上的显著优势。➡
RC48的三线治疗效果甚至能够媲美二线标准治疗方法的临床效果,且安全性良好,表明其具有二线治疗潜力。➡
国内RC48研发进度大幅领先于其他在研胃癌ADC药物。
3.2.4. RC48治疗尿路上皮癌的竞争格局:
核心观点:尿路上皮癌(UC)是RC48的又一核心适应症,我们预计公司将于今年第三季度向中国CDE报NDA,2022年获批进入尿路上皮癌二线治疗。此外RC48已获得FDA授予的尿路上皮癌适应症突破性疗法资格,2021年在美国启动II期临床试验。RC48在HER2过表达晚期尿路上皮癌中的临床数据优异,相比已上市竞品,大幅提高患者的缓解率和生存期,我们认为RC48将成为UC二线治疗中目前全球最好的治疗药物。小样本的临床数据显示,在HER2低表达晚期UC患者中也同样具有显著的治疗效果,有望成为晚期尿路上皮癌治疗的突破性产品。
尿路上皮癌流行病学特征:尿路上皮癌主要由膀胱癌组成(占比90%),根据Globocan 2020年的统计,中国及美国每年新发膀胱癌患者人数分别为8.57万人、8.06万人,五年累计患病人数约为23.54万人、26.92万人,并以约3%的速度逐年增加。弗若斯特沙利文统计,约48%的尿路上皮癌患者存在HER2过表达,20%患者HER2低表达。 晚期尿路上皮癌目前的治疗手段及二线后用药方案:晚期尿路上皮癌一线治疗以化疗为主,二线及之后的治疗优先考虑参加新药临床试验以及采用免疫疗法,其中PD-1抗体是主要的治疗方式。多个尚未在国内获批的药物也被作为备选治疗方案,出现在2020年CSCO尿路上皮癌的诊疗指南中。含铂化疗是晚期UC的有效一线疗法,但由于化疗的耐药性,患者的PFS和OS较短,一线治疗后仅有25%~55%的患者能接受二线治疗。
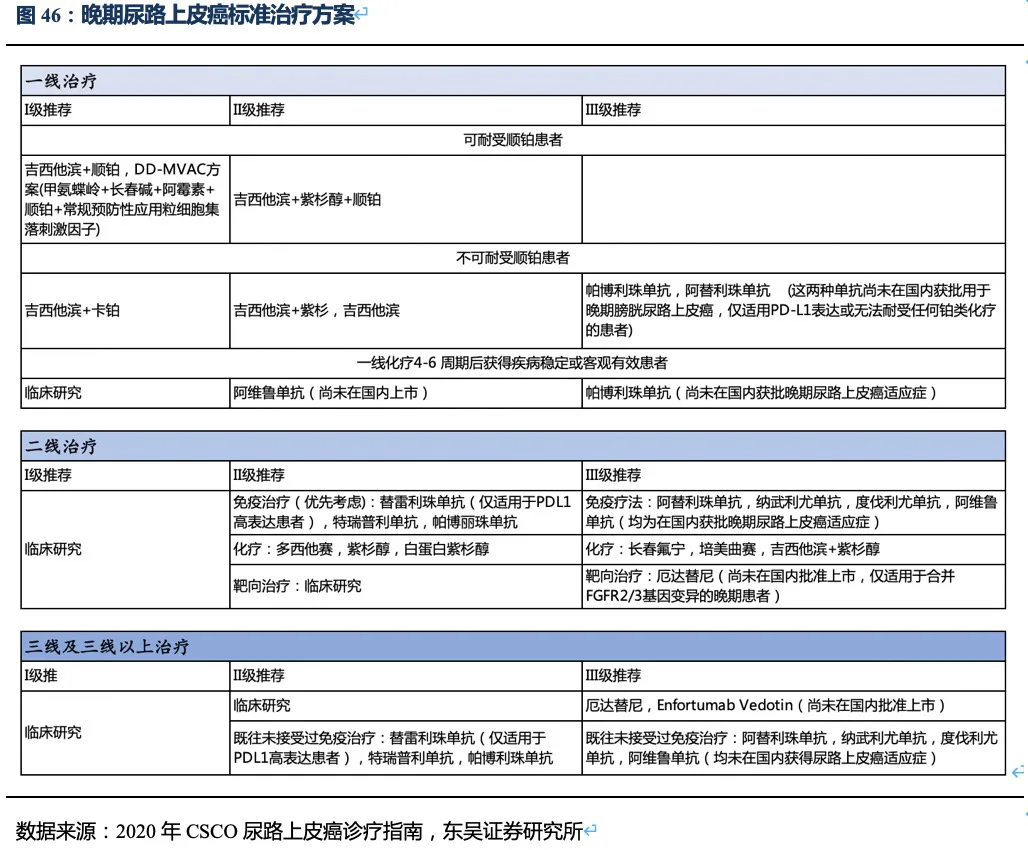
已获批上市的晚期尿路上皮癌药物:在我国,获批治疗尿路上皮癌治疗的靶向药只有百济神州的替雷利珠单抗(二线疗法)。此外,君实在2021年4月公布其特瑞普利单抗二线治疗UC的适应症获NMPA批准上市。其他多种PD-(L)1单抗均在美国FDA批准用于UC,而在我国处于临床III期阶段尚未获批。罗氏阿替利珠单抗和阿斯利康的度伐利尤单抗分别于今年的3月和2月主动撤回UC适应症,主要原因是以II期数据有条件上市批准后,III期验证性临床试验未达到主要终点。Enfortumab
vedotin(靶向细胞骨架蛋白Nectin-4的ADC药物)是目前全球已上市二/三线治疗效果最好的上市药物之一,目前已在国内获批进行临床试验。
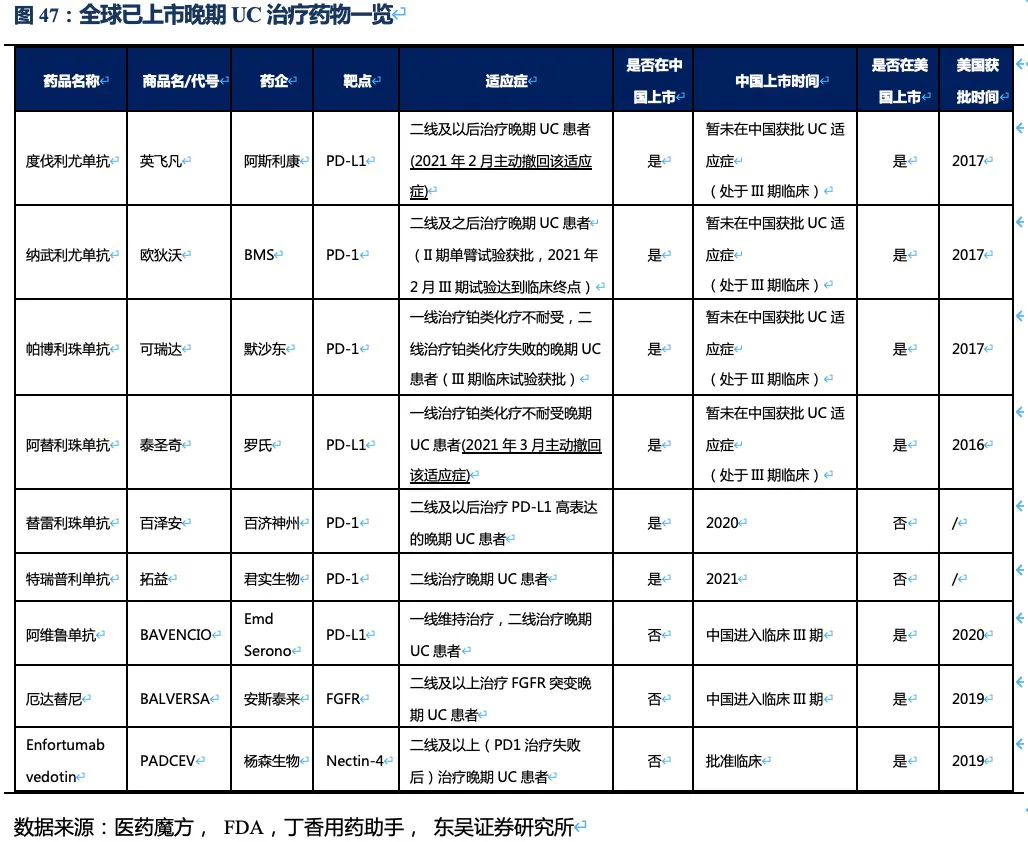
比较已上市的UC治疗药物,Enfortumab vedotin、特瑞普利单抗和厄达替尼应该是目前二线及以上治疗药物中有效性最高的三种药物,分别代表三种不同的治疗机制(ADC、肿瘤IO治疗、小分子靶向治疗)。根据2021年3月ASCO会议上报道的全球II期临床试验EV-201的数据,Enfortumab vedotin在二线和三线治疗中的ORR分别达52%和40.6%。特瑞普利单抗在二线治疗中的ORR达25.8%、mOS为14.4个月,尤其对于PD-L1阳性患者,ORR达41.7%、mOS达到35.6个月。
上述三种药物二线治疗的有效性与已经获批的一线用药帕博利珠单抗相当。而PD-(L)1单抗对二线及以上UC患者的ORR一般在15%~25%左右。
随着RC48以II期临床数据获批上市,我们认为RC48将成为UC二线治疗中目前全球最好的治疗药物。荣昌生物的RC48尿路上皮癌适应症目前正处于临床III期,II期数据显示RC48对HER2+
UC患者ORR可达51%,DCR可达90%,mPFS超过6.9个月,在PD-1单抗治疗失败的患者中ORR达到75%,优于目前所有在中美获批的UC
二线及以上的治疗药物。此外RC48还在HER2低表达UC患者中开展临床试验(II期),初步的临床结果显示出RC48的治疗潜力。RC48无疑将成为晚期尿路上皮癌治疗的突破性药物,期待后续更多数据显示出RC48优异的治疗效果。
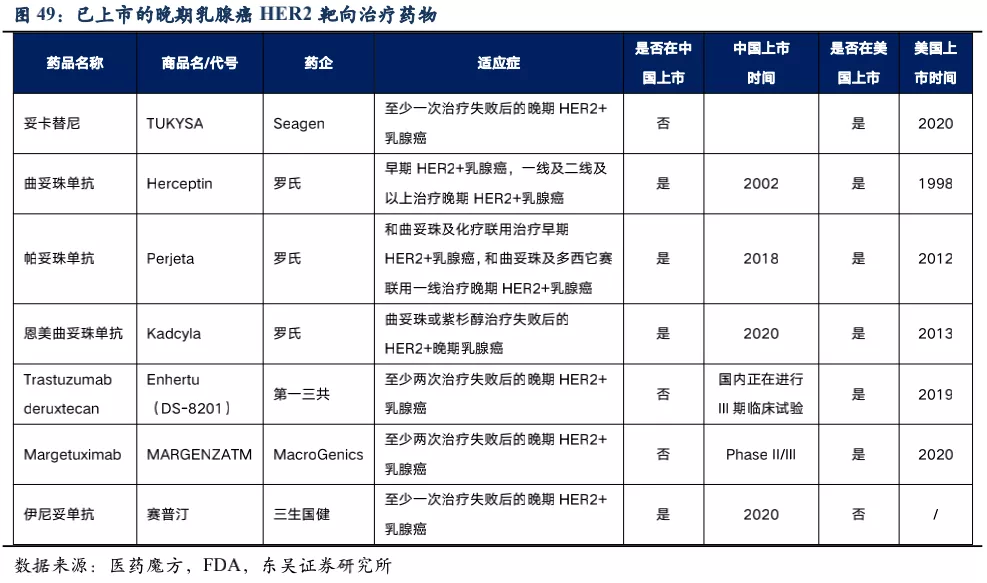
3.2.5. RC48治疗HER2低表达乳腺癌的竞争格局
核心观点:乳腺癌患者众多,但HER2阳性乳腺癌赛道竞争激烈,荣昌生物重点推进RC48在HER2低表达乳腺癌患者中的临床试验,目前已进入关键III期临床,我们预计2022年申请NDA。从安全性和有效性两个方面看,我们认为RC48针对HER2低表达乳腺癌适应症相对DS-8201具有一定的竞争潜力,RC48安全性好于DS-8201,在I期临床试验中HER2低表达患者(n=48,给药剂量为2.0mg/kg)ORR=39.6%,有效性与DS-8201相当,而安全性优于DS-8201。 乳腺癌流行病学特征:乳腺癌是女性最常见的癌症,根据Globocan 2020年统计数据,我国每年新发乳腺癌患者人数为41.6万人,5年患病人数达139万,并以约1.5%的速度逐年增加。在对乳腺癌进行诊断时,需要确定其分子分型,一般需要检测HER2、ER(雌激素受体)、PR(孕激素受体)的表达,乳腺癌根据这些受体的表达与否进行分子水平上的细分,决定了后续患者的用药方案。
HER2低表达乳腺癌具有异质性,包括HR阳性型(包括ER和/或PR阳性)和三阴性乳腺癌。乳腺癌是HER2靶向药的主要适应症之一,这些靶向药大多针对HER2高表达患者。根据美国NCCN指南79%的乳腺癌患者为HER2阴性,这其中超过一半为HER2 低表达(占整个乳腺癌的比例为40%~50%)。对于这部分HER2低表达患者,可能存在着部分患者HR受体也为阳性,但由于目前临床中对于内分泌治疗获益的HR cut-off值也存在分歧和争议,因此在后续表3的计算中,我们按25%计算HER2低表达人群中使用RC48-ADC获益的人群。 已上市的晚期乳腺癌HER2靶向治疗药物:目前已上市的HER2靶向药物,均针对HER2高表达(IHC 3+或FISH+)乳腺癌患者,对HER2低表达(IHC 2+/FISH-或IHC 1+)乳腺癌患者并未覆盖,RC48避开了竞争最为激烈的领域,竞争格局相对较好。
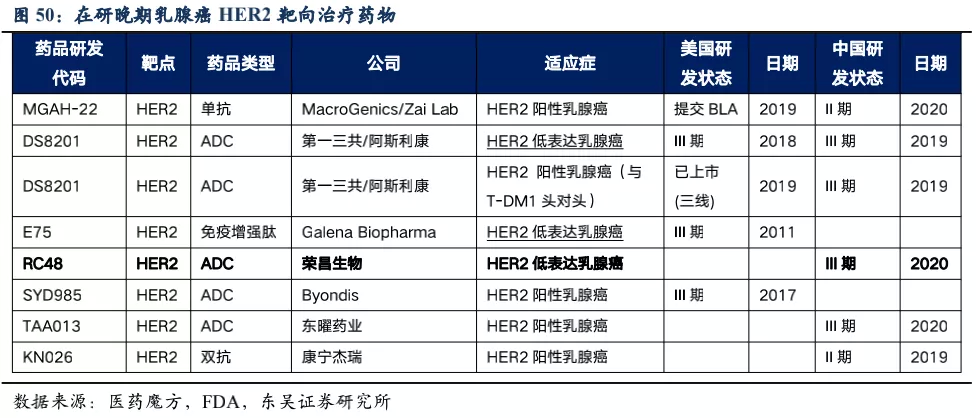
在研晚期乳腺癌HER2靶向治疗药物:HER2低表达乳腺癌治疗领域在研竞品中,RC48将面临来自第一三共/阿斯利康的产品DS-8201(Trastuzumab deruxtecan)的竞争。但考虑到DS-8201因严重不良反应存在FDA黑框警告,具有较高的间质性肺病的发生率以及出现患者死亡的可能性,安全性将会是RC48相对DS-8201的优势之一。
根据2020年J Clin
Oncol杂志数据,DS-8201针对HER2低表达乳腺癌的54例Ib期临床试验数据看(见RC48胃癌分析部分图表),≥5线治疗的的患者数占83.3%,患者整体的ORR为37%、DCR为83.3%,mPFS为10.4个月,mOS为29.4个月。亚组分析看,5.4mg/kg组(n=21)
ORR=33.3%,6.4mg/kg组(n=33)ORR=39.4%;IHC2+组(n=26)ORR=38.5%,IHC1+组(n=28)
35.7%;之前用过HER2靶向治疗(n=10)ORR=30.0%,之前未用过HER2靶向治疗(n=44)ORR=38.6%;日本人组(n=27)ORR=44.4%,美国人组(n=27)ORR=29.6%。安全性方面≥3级以上不良反应发生率为63%,20.4%的患者因不良反应停药,22.2%因不良反应而降低了用药剂量。因此,从安全性和有效性两个方面看,我们认为RC48针对HER2低表达乳腺癌适应症相对DS-8201具有一定竞争潜力,在所有患者中(n=48,给药剂量为2.0mg/kg)RC48的ORR=39.6%、mPFS=5.7个月,IHC2+(n=35)ORR=42.9%、mPFS=6.6个月,IHC1+(n=13)ORR=30.8%、mPFS=5.5个月。
3.2.6. 差异化布局的ADC平台领头羊,预计2030年RC48销售额近24亿人民币
由于大量实体瘤内浸润的淋巴细胞水平不足,免疫疗法经常无法达到理想的治疗效果,且肿瘤细胞容易通过受体内化等方式产生对特定单抗药物的耐药性,以上原因导致单抗在实体瘤上的治疗效果受限,作为新型的靶向用药ADC药物将在实体瘤领域大放异彩。除了RC48,荣昌生物还有另外三款ADC药物,分别为靶向间皮素的ADC——RC88(可用于卵巢癌、胆道癌等),靶向c-MET的ADC——RC108(可用于非小细胞肺癌等),以及未知靶点的ADC——RC118,三款ADC药物均用于治疗实体瘤。借助良好的安全性,公司的ADC管线还可以和PD-(L)1单抗联用,助力ADC品种治疗线数前移。 除了上述三个在三年内可以获批上市的适应症,RC48也在积极推进非小细胞肺癌及胆管癌的临床试验。若仅测算国内市场的三个适应症(胃癌、尿路上皮癌和HER2低表达乳腺癌),我们预计2030年RC48将在国内实现近24亿人民币的销售额。
我们对RC48的价格预期参考已上市的HER2-ADC恩美曲妥珠单抗(T-DM1),假设患者平均体重60kg,患者推荐用药剂量为3.6mg/kg, 每次需使用216mg即2支100mg, 每21天注射一次,一年需要使用14个周期,恩美曲妥珠单支价格为19282元/100mg/支,一年治疗费用为54万。为了充分发挥RC48的先发优势,我们预计其定价大约为T-DM1的35%,即上市初定价为18.9万元/年,医保谈判后价格将降低50%,最终年用药金额为9.45万元。我们预计上市首年慈善赠药后的年用药费用和医保覆盖后的费用差距不大。
根据《柳叶刀》2018年数据,我国胃癌5年生存率不到20%,存量市场相对较大,因此对销售额测算时以存量患者作为基数。尿路上皮癌约60%是浸润性肿瘤,预后不佳,在我国5年生存率不超过5%,此处测算仍以存量患者作为预测基数。我国乳腺癌患者5年生存率高达83.2%,因此在市场测算中,也以存量患者作为测算基数。渗透率随着第一年进医保后,次年渗透率增长2~3倍,以后每年渗透率的增长幅度假设为10%~20%。
此外,还需要说明的是,在胃癌市场的测算中,我们考虑了二线治疗患者人群,原因在于:一方面胃癌缺乏有效的治疗药物,安全性良好的RC48是存在超适应症用药的情况,另一方面RC48与PD-(L)1联合治疗线数前移的可能性较大,且存在研究者发起的临床试验数据作为支持。
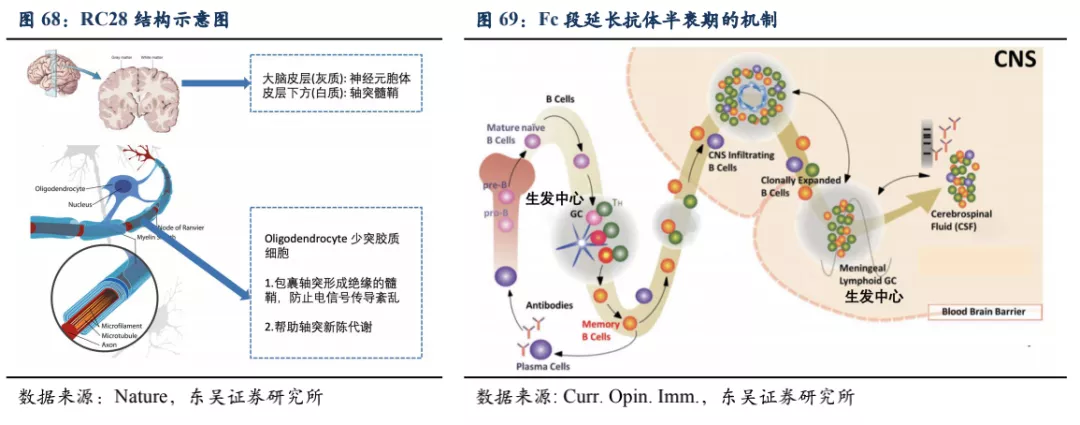
4. RC28:黄斑病变等眼科疾病下一代重磅产品
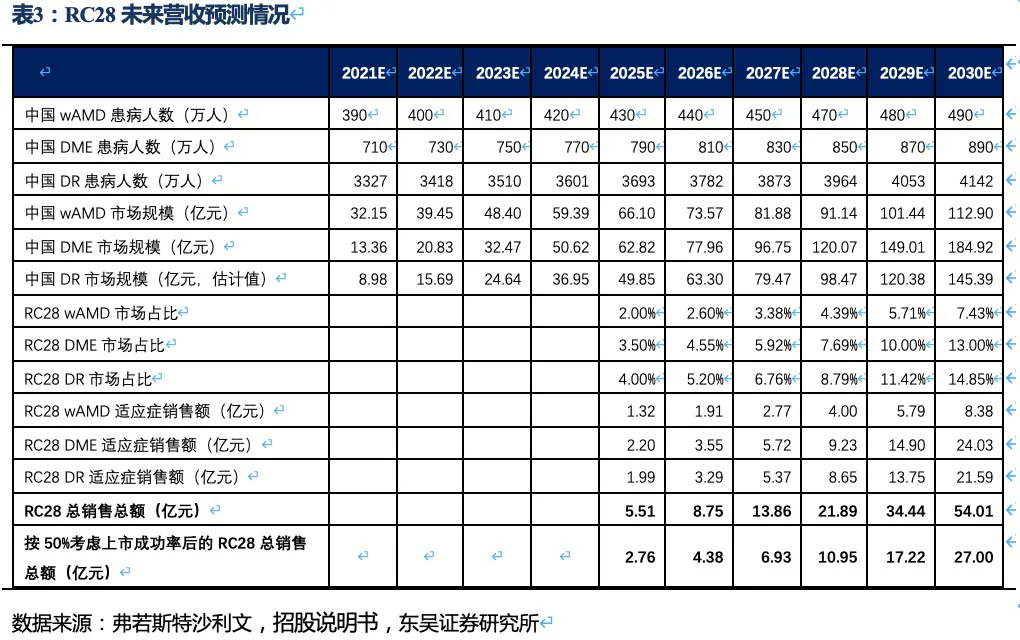
核心观点:眼科药物RC28是一种同时靶向血管内皮生长因子(VEGF)和成纤维细胞生长因子(FGF)的双靶点融合蛋白,糖尿病性黄斑水肿(DME)和糖尿病视网膜病变(DR)两个适应症均于2021年进入II期临床,老年性湿性黄斑病变(wAMD)适应症预计也在今年进入II期临床。作为下一代差异化的双靶眼科用药,RC28在DME和DR两个适应症竞争格局良好且研发进度居第一梯队,我们预计RC28将于2024年~2025年在国内获批上市,在考虑上市成功率和降价预期的情况下,2030年销售额预计将达到27亿元。
4.1. 黄斑病变眼科用药市场空间较大
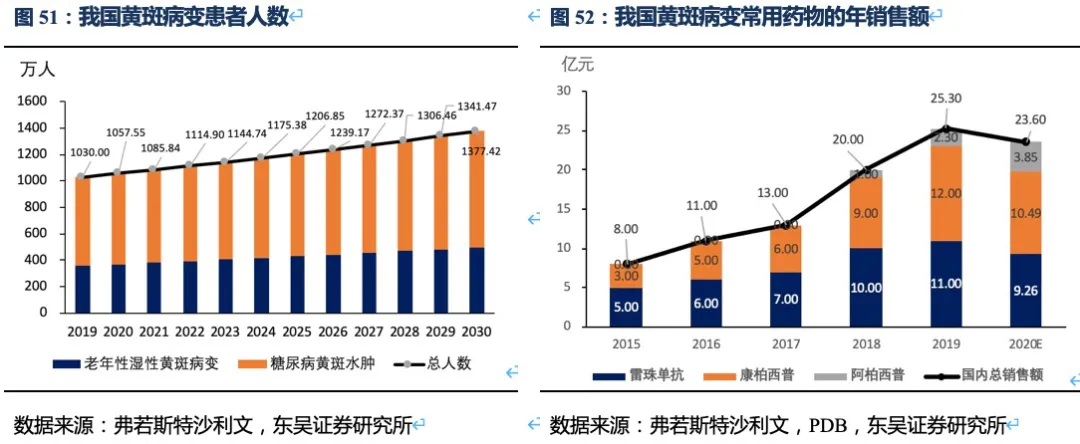
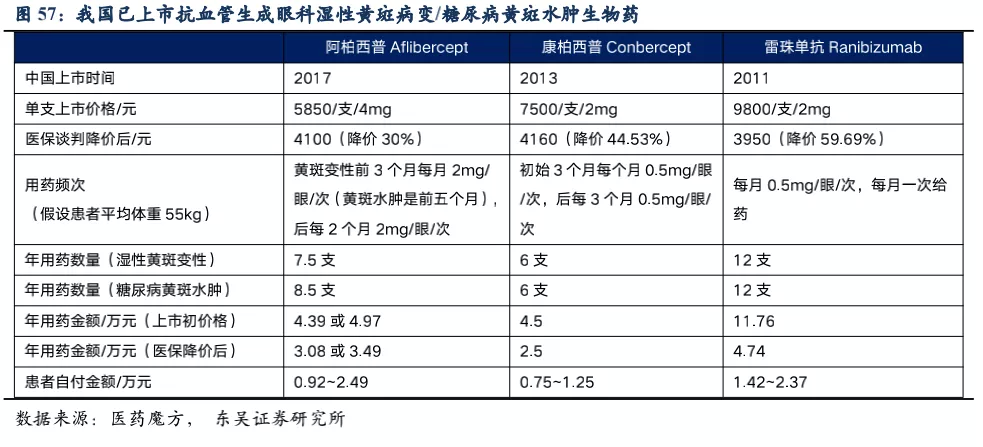
RC28的适应症包括老年性湿性黄斑病变、糖尿病黄斑水肿和糖尿病视网膜病变。A. 老年性湿性黄斑病变(wAMD)是由于眼部异常的新生血管在视网膜下生长,引起视网膜出血、水肿以及组织破坏而导致的。B. 糖尿病黄斑水肿(DME)是由于患者视网膜组织分泌过量的血管内皮细胞生长因子(VEGF)和促炎性细胞因子,从而导致血管结构和通透性改变,血-视网膜屏障破坏,造成视网膜黄斑中心肿胀。C. 当血管损伤和新血管形成导致血液和/或体液渗出到视网膜时,就会发生糖尿病性视网膜病变(DR)。血管生成抑制剂是治疗湿性黄斑病变、糖尿病黄斑水肿的主要药物,患者治疗不及时会导致永久性视觉损伤甚至失明。我国是眼科疾病大国,根据弗若斯特沙利文统计,我国黄斑病变的发病率几乎是美国的3倍,我国湿性黄斑病变人数360万人、糖尿病黄斑水肿人数670万人,并以每年2.5%-3%的速度逐年增加,存量患者数超千万。我国已获批用于黄斑病变的药物包括雷珠单抗、阿柏西普、康柏西普,2020年这三种药物的销售总额大约23.6亿人民币,近5年的复合增速为24.2%。
4.2. RC28双靶点抑制血管生成治疗效果更显著
4.2.1 RC28的作用机制和结构优势
RC28为双靶点融合蛋白,其一端为血管内皮生长因子受体(VEGFR)以及成纤维细胞生长因子受体(FGFR) 的配体结合域,这些结构能够高效结合VEGF和FGF。单靶点抗VEGF疗法面临的主要挑战之一是当VEGF被抑制时,其他促血管生成因子(如FGF-2)的表达上调。RC28另一端为抗体的Fc段,带有Fc段的蛋白药物在被细胞吞噬内化后通过结合细胞内的Fc受体避免被降解,并可以通过细胞膜融合再回到细胞外环境中,被重新利用从而延长药物在血清中的半衰期、减少患者的用药频率,对于需要直接注射入眼内的药物,长半衰期能够极大提高患者的依从度。
已知FGF和VEGF是血管内皮细胞生长增殖以及血管生成过程中重要的促进因子,RC28双靶点抑制剂能够更有效地抑制血管生成。体外的实验显示,RC28对于血管内皮细胞增殖的抑制性是最强的(相对康柏西普和阿柏西普等),提示RC28可能具有更优的治疗作用。
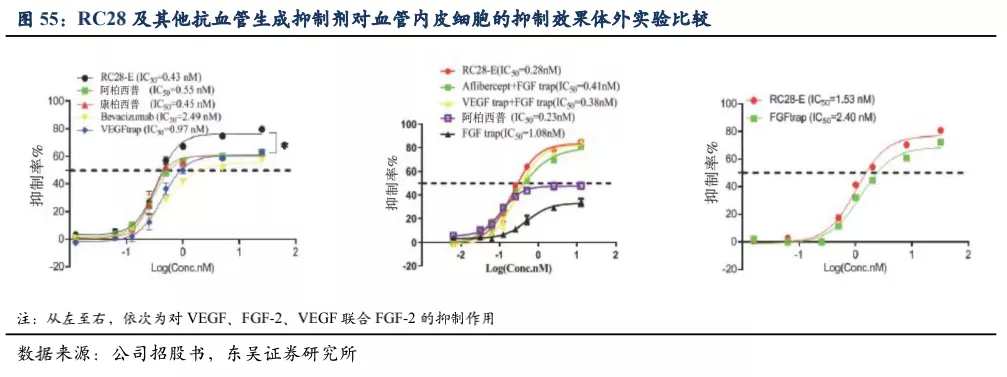
4.2.2 RC28的竞争格局分析
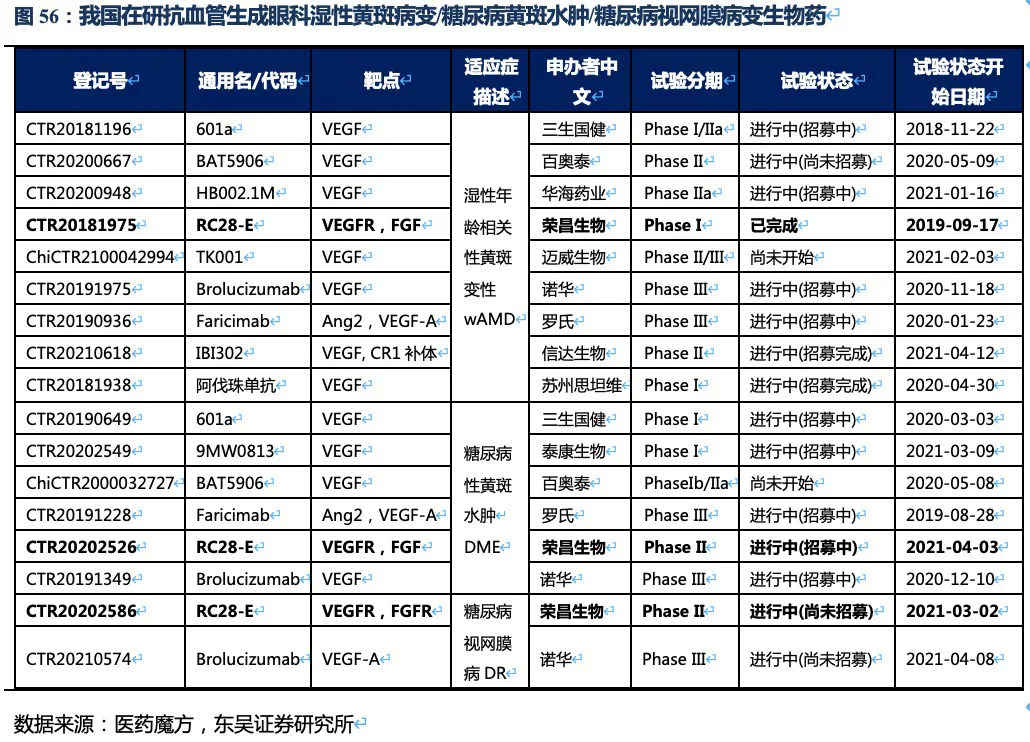
目前国内上市的3种抗血管生成黄斑病变生物药均是单靶点药物。除去一些中药等口服制剂,仅看国内在研生物药竞争较为激烈,wAMD适应症大部分处于临床II期或III期,而DME和DR两个适应症竞争格局较好,且荣昌生物的RC28的进度略落后于罗氏或诺华。
从在研品种看,罗氏、信达生物和荣昌生物均为双靶点生物药,其中罗氏的Faricimab对于wAMD和DME两个适应症的共四项全球III期临床试验在2020年底达到了主要临床终点(与已获批的VEGF抑制剂相比非劣效性标准)。且试验组中有一半左右的受试者在第一年治疗时,治疗的间隔时间延长到16周(其他受试者给药间隔为8周或12周,对照组为每8周给药一次阿柏西普),这是治疗眼科黄斑病变在研药物中首次在III期试验中达到4个月的持久性。
4.3. 新一代双靶长效眼科用药,预计2030年RC28的销售额达到27亿元
从罗氏的Faricimab等在研品种看,竞品间药效差别不会很大,双靶点药物的好处是降低复发的可能性、减少长期用药可能导致的纤维化,但关键在于延长给药周期,降低患者眼部注射的不适感,根据康柏西普的给药频率推测,我们预计RC28的注射频率为3个月左右。用药金额经过2019年医保谈判后,年用药金额也基本降至2.7万元左右。
根据弗若斯特沙利文的数据以及我们的测算,以wAMD、DME和DR三个市场未来的规模及RC28的市占率测算出未来的营收规模。由于DME和DR的市场竞争格局相对较好,因此市占率假设也相对较高。最终,考虑上市成功率及后续竞品降价的可能性,我们预计RC28在三个眼科适应症的销售额合计约27亿元左右。
5. 盈利预测与估值评级
5.1. 盈利预测:预计公司2030年营收将超过164亿人民币
风湿免疫疾病领域:泰它西普是目前全球所有已上市以及在研产品中临床治疗效果最好的SLE治疗药物,打破长久以来SLE患者无有效药物可用的局面,且后续尚有其他六个在研的风湿免疫病适应症,我们预计也将在2023~2024年获批上市。无论国内还是海外风湿免疫病患者数量庞大,未满足临床需求长久存在。随着我国风湿免疫科的逐步发展,医生、患者对生物药优势的认知提高及人均支付水平的提升,泰它西普百亿销售额指日可待。
肿瘤领域:作为新一代的肿瘤免疫治疗药物ADC在多种实体瘤的优效得到了充分的验证。荣昌生物作为国内最领先的ADC药物研发平台,差异化的临床布局抢占了先发优势,RC48在胃癌和尿路上皮癌已验证了有效性,预计将在2021~2022年在国内及海外获批上市。后续涉及的适应症还包括HER2低表达实体瘤以及HER2阳性胆管癌和非小细胞肺癌。靶向c-Met和间皮素的ADC药物也已进入临床I期,同时预计今年也会有双抗品种进入临床阶段。根据我们的测算RC48在胃癌、尿路上皮癌,以及HER2低表达乳腺癌三个适应症2030年的销售额将接近24亿元。
眼科疾病领域:眼科领域是肿瘤和自免外又一重磅赛道,RC28是荣昌生物自研的双靶点融合蛋白,糖尿病性黄斑水肿(DME)和糖尿病视网膜病变(DR)两个适应症均于2021年进入II期临床,老年性湿性黄斑病变(wAMD)适应症预计也在今年进入II期临床。作为下一代差异化的双靶眼科用药,RC28在DME和DR两个适应症竞争格局良好且研发进度居第一梯队,我们预计RC28将于2024年~2025年在国内获批上市,在考虑上市成功率和降价预期的情况下,2030年销售额预计将达到27亿元。
根据前文对药效、竞争格局、市场空间的分析,我们预计荣昌生物三大核心产品泰它西普(RC18)、RC48和RC28的销售额在2030年将超过164亿人民币。其中泰它西普将为公司贡献大部分营收,特别是泰它西普的海外市场收入将成为公司营收的重要组成部分,随着IgA肾炎等适应症在海外获批,海外销售额很可能超过国内空间。
5.2 估值评级
我们预计2021~2023年公司实现收入3.10、9.80、22.59亿,对应归母净利润-8.20、-6.19、-1.32亿。根据测算,我们预计公司将于2024年实现盈利,2025年归母净利达到8.77亿,对应PE 49倍。以PS计算,2021~2025年收入对应的PS分别为138、44、19、17和10倍。
A. 对于具有明显先发优势和技术壁垒的平台型公司,由于三大核心品种均有较高的销售峰值,按PS估值法,2030年可以给予10倍PS,即对应2030年市值1647.8亿人民币,按10%折现后对应2021年目标市值635.3亿人民币(即738.7亿港币,人民币兑港币汇率按1:0.86计算)。
B. 根据三大品种的未来销售峰值,即使2021年给予4倍PS,2021年目标市值也可以达到659.1亿人民币(766.4亿港币)。
因此,综上从PS估值角度年内或有较大上涨空间,以两年维度看我们认为荣昌生物是一家市值千亿的平台型公司,首次覆盖给予“买入“评级。
6. 风险提示
国内风湿免疫病患者对生物药物的认知和接受需要一个过程,这会影响到泰它西普的推广效果和销售放量,因此在部分年度的销售额可能不及我们的预期。
核心产品临床实验结果不及预期,如泰它西普或RC48或RC28等在研产品的临床试验仍在推进当中,尤其海外临床试验存在较多不受控因素,相关临床试验进度和结果存在不及预期的风险。
同类产品竞争风险,特别是RC48-ADC药物和RC28眼科用药随着越来越多竞品上市,市场竞争加剧,可能导致相关产品销售额不及预期。
港股交易风险,由于港股没有涨跌停限制,单日波动较大等使得港股交易存在较大风险。
7. 附录
7.1. 公司基本概况
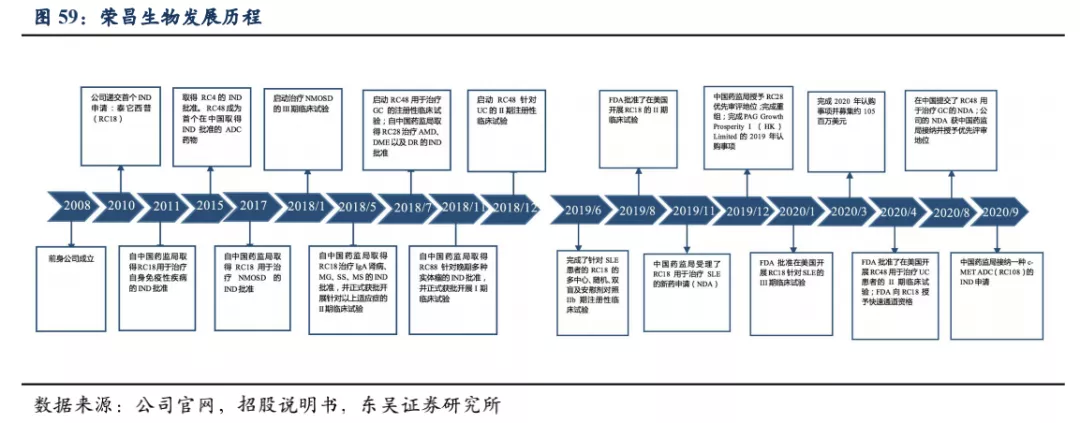
7.1.1. 公司发展历程
荣昌生物成立于2008年,是一家正在进入商业化阶段的平台型生物制药公司,致力于发现、开发和商业化创新的、有特色的生物药,用于治疗中国乃至全球多种未被满足的风湿免疫、肿瘤和眼科疾病类医疗需求。
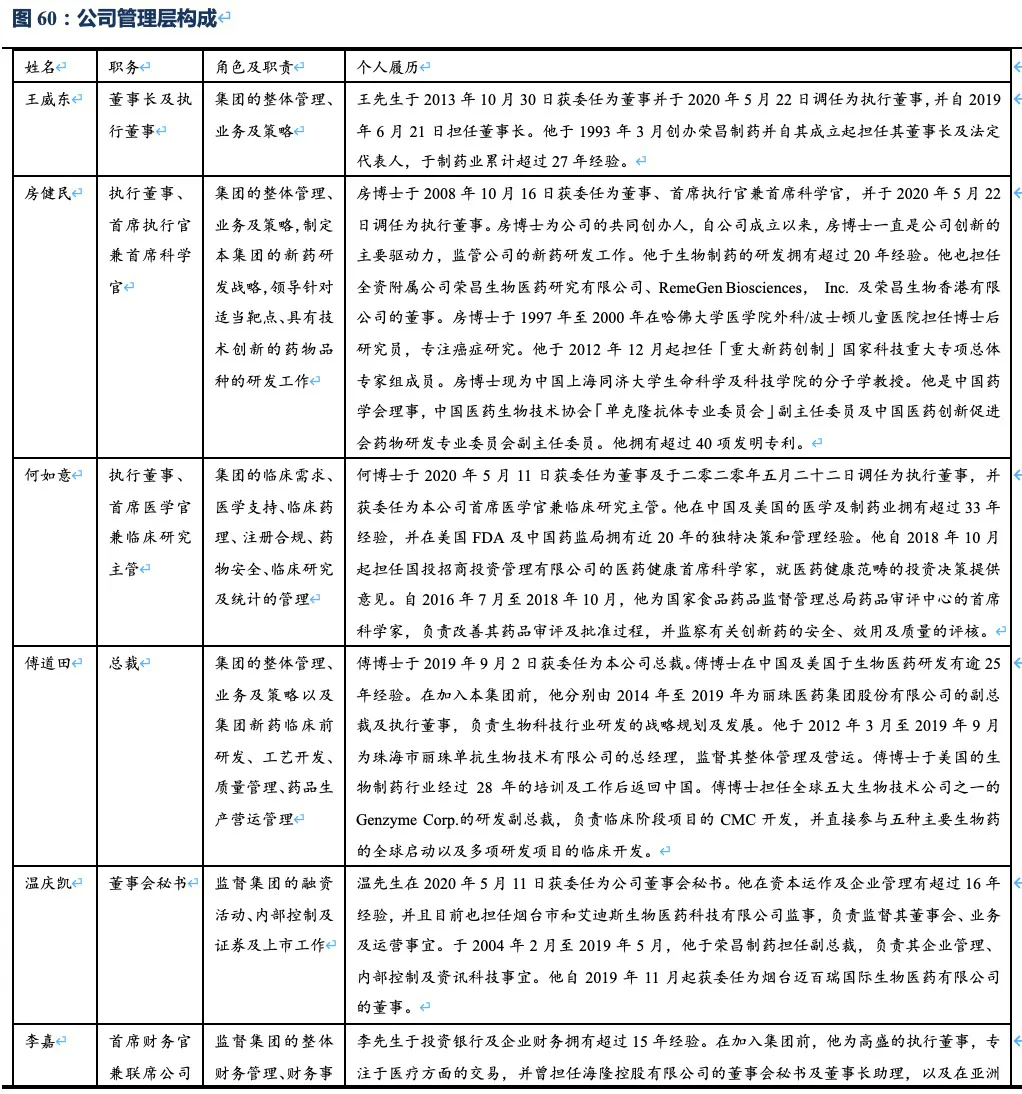
7.1.2. 公司的管理层构成
公司组建了一支经验丰富且具有国际视野的高级管理团队,成员平均拥有逾20年的行业经验,并在创新药物开发、临床开发及商业化方面拥有成功经验,具备全球化竞争能力。公司的联合创始人、首席执行官兼首席科学官房健民博士,是国内生物制药行业为数不多的、在生物创新药从发现到开发再到商业化的整个流程方面拥有成功往绩记录的企业创始人,是三款核心产品RC18、RC48和RC28的发明者。他曾经在哈佛大学医学院担任博士后研究员专注癌症研究,现在公司担任执行董事、首席执行官兼首席科学官,于生物制药的研发拥有超过20年经验和超过40项发明专利。 何如意博士现任公司执行董事、首席医学官兼临床研究主管。他曾在美国FDA和中国药监局(CDE)拥有近20年的决策和管理经验,积累并熟知了中国、美国及其他地区的监管审查规定及审批程序的大量专业知识。
7.2 其他风湿免疫疾病的背景介绍
7.2.1 系统性红斑狼疮疾病活动程度评分标准
系统性红斑狼疮患者疾病严重程度按照国际通用的SLEDAI-2K标准衡量,具体评分标准如下:

7.2.2. 视神经脊髓炎疾病疾病特征介绍
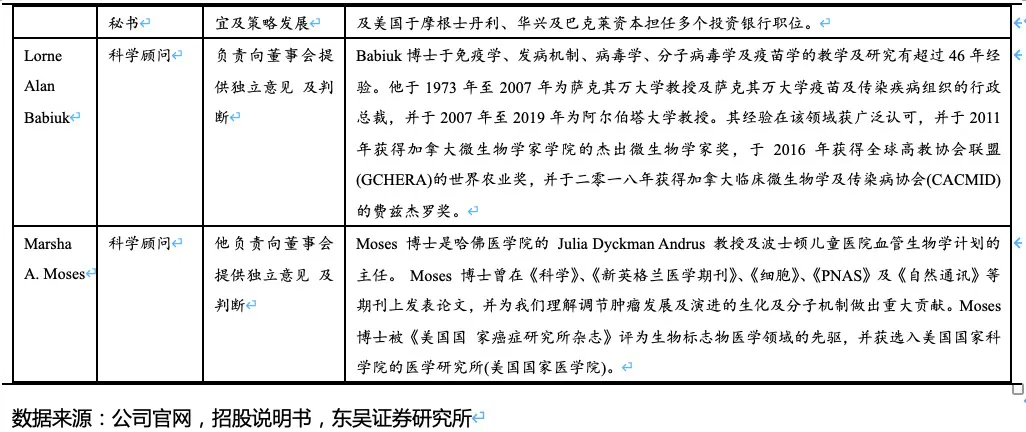
视神经脊髓炎(NMO)是一种免疫介导的以视神经和脊髓受累为主的中枢神经系统(CNS)炎性脱髓鞘疾病。患者体内会出现大量水通道蛋白4抗体(AQP4-IgG),AQP4是中枢神经系统中最多的水通道蛋白, 患者体内产生的自身抗体攻击AQP 4从而造成该蛋白水平降低,神经细胞遭到破坏, 患者正常的神经冲动传导受损。NMO临床上多以严重的视神经炎(ON)和纵向延伸的长节段横贯性脊髓炎(LETM)为特征表现,常于青壮年起病,女性居多,复发率及致残率高。
7.2.3. 类风湿关节炎疾病特征介绍
类风湿性关节炎(RA)是一种慢性系统性风湿免疫疾病。其成因为机体自身的免疫系统对关节组织及细胞错误地进行攻击,从而导致炎症以及关节滑膜(synovium)的增厚,破坏关节处的软骨及骨组织。同时,肌腱及其他保持关节连接的组织会变松散而被拉伸,导致关节处变形, RA 多侵犯手指、膝盖等关节, 且损伤一旦形成即不可逆, 患者生活质量受到严重影响。此外作为一种系统性风湿免疫疾病, RA患者身体多个组织如关节、皮肤、眼睛、肺、心脏和血管等均会发生损伤。
7.2.4 IgA 肾炎疾病特征介绍
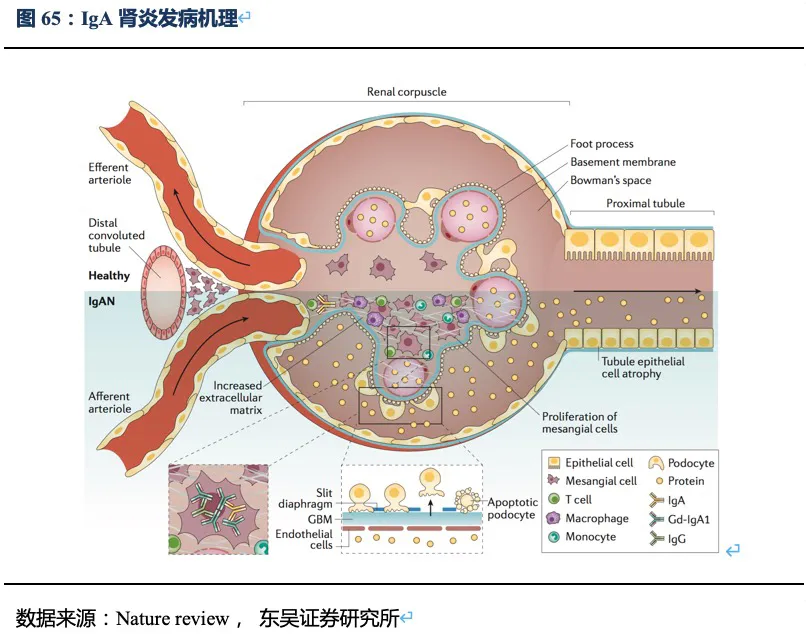
IgA肾炎(IgA nephropathy):是全世界最常见的原发性肾小球肾炎,这是一种自身免疫性疾病,表明免疫抑制治疗可能潜在地有助于临床缓解。IgA肾病患者在肾小球膜中有IgA抗体呈颗粒状的沉积。这是由于两种免疫球蛋白分支之一的IgA1,一部分位于特殊的富脯氨酸铰链区的丝苏氨酸O-端糖基化的蛋白分子异常。这些糖基缺失导致组织中的IgA分子聚合,特别是在肾小球膜。IgAN表现为复发性血尿和/或蛋白尿,通常在非特异性的上呼吸道感染以后就会出现,而链球菌性的肾小球肾炎则通常在原发性感染后数周才会出现血尿;少数情况下胃肠道感染或尿道感染也可以称为诱发因素。所有这些炎症都会激活黏膜反应,从而产生IgA抗体。随着研究的进展发现,IgAN的自然病程为非良性,并且可能导致肾功能的严重恶化。大约20-40%的IgAN患者将在10-20年内发展为终末期肾脏疾病(ESRD),或需要进行肾脏替代治疗。大量的全身性疾病都和IgA 肾病有关,比如肝脏衰竭,腹腔疾病,关节炎,Reiter综合征, 强制性脊椎炎和HIV。因此,寻找预防患者肾脏衰竭的最佳策略至关重要。
7.2.5. 干燥综合症疾病特征介绍
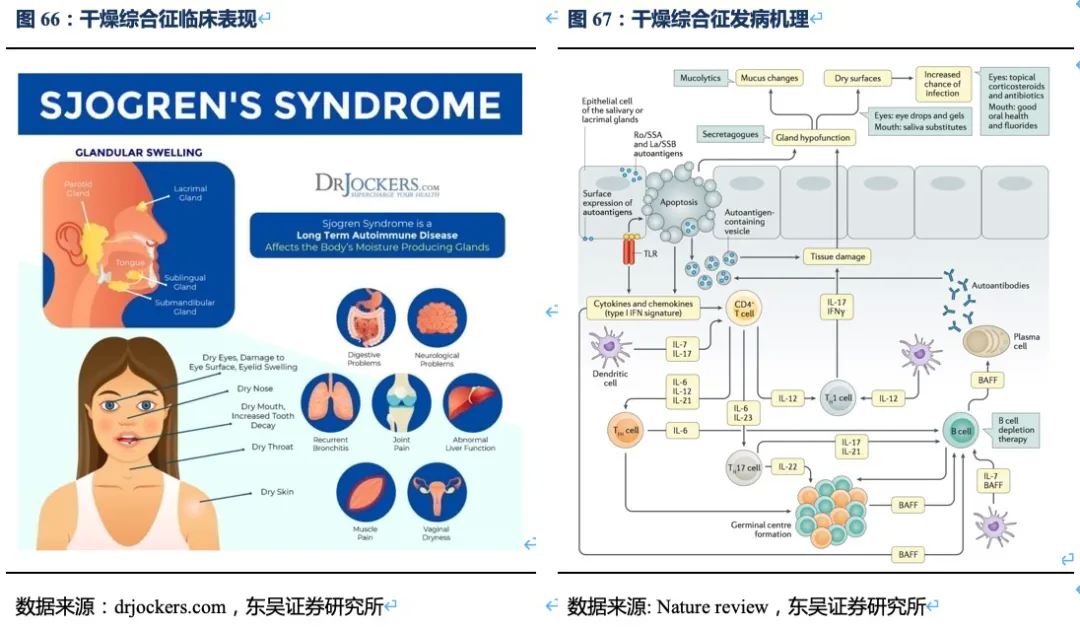
干燥综合征(SS):是一个主要累及外分泌腺体的慢性炎症性自身免疫病,又名自身免疫性外分泌腺体上皮细胞炎或自身免疫性外分泌病,该病的患者其自身的免疫系统错误的对自身功能正常的细胞进行攻击,并引发炎症。临床表现分为局部唾液腺和泪腺受损功能下降而出现口干、眼干外,和系统性由于外分泌腺及腺体外其他器官的受累而出现多系统损害的症状,包括关节炎、肾炎、血细胞减少、肺炎和血管炎。SS的发病机制是多步骤过程,在遗传易感个体中由环境因素(最可能是病毒)触发从而激活黏膜上皮细胞。该过程导致固有免疫和适应性免疫系统之间持续相互作用刺激自身反应性B细胞,产生自身抗体,构成免疫复合物,扩大α干扰素的生成,引起免疫系统激活的环路,并导致唾液腺、泪腺及其他组织的慢性炎症。出现腺体外表现的原因可能包括:自身免疫性外分泌腺体病,类似于唾液腺的病变,比如间质性肾炎、胆汁性胆管炎;免疫复合物沉积,比如冷球蛋白血症性血管炎;结外淋巴细胞增生,比如淋巴细胞型间质性肺炎。靶组织内B细胞受到长期刺激可能促进淋巴瘤生成。
7.2.6. 多发性硬化症疾病特征介绍
多发性硬化症(MS):是一种中枢神经脱髓鞘疾病。大脑内神经元拥有长长的轴突负责神经电信号的传导,轴突外侧被少突胶质细胞包裹形成髓鞘,帮助轴突代谢。髓鞘的另一个重要功能类似电线的绝缘层,防止神经信号传导的紊乱。而MS患者大脑内的髓鞘受到免疫系统的攻击被破坏(大脑内的自身反应性B细胞有2个来源:一种是从外周血中进入,一种是在大脑中扩增产生的,抗 B细胞增殖是治疗MS的潜在机制),电信号传导紊乱,造成一系列严重的疾病。MS患者会出现运动、感觉神经异常,造成肢体的瘫痪,视觉听觉障碍等。
7.2.7 重症肌无力疾病特征介绍
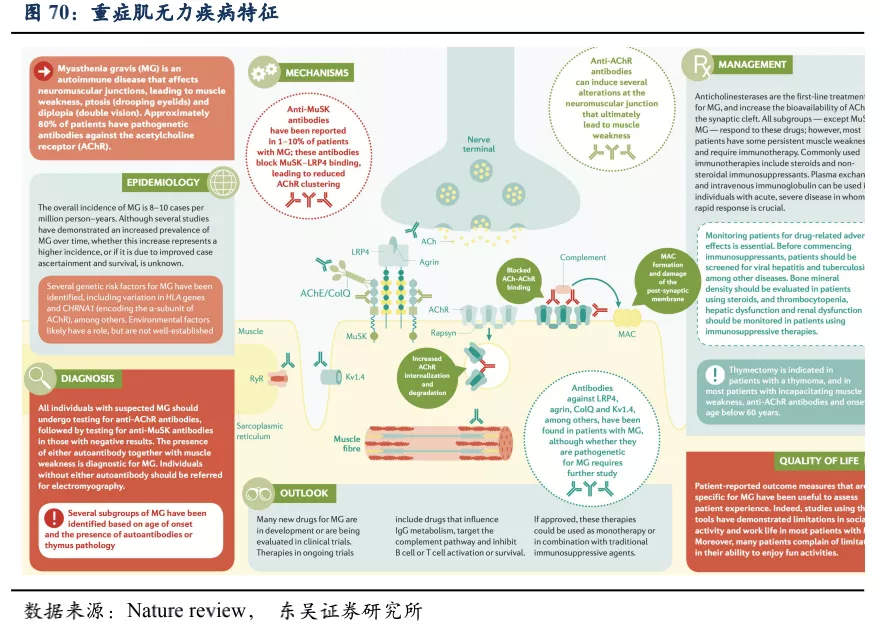
重症肌无力也是一种风湿免疫疾病, 患者体内产生大量针对乙酰胆碱受体(AchR)的自身抗体。Ach-AchR的相互作用控制着神经-肌肉接头的信号传导, 确保肌肉能够按照神经信号指示完成收缩和舒张, 维持人体正常运动。重症肌无力患者体内, 大量的自身抗体和 AchR 结合, 使得 Ach 无法顺完成信号传导, 肌肉无法正常收缩, 从而造成患者肌无力、眼睑下垂、视物模糊、易疲劳、咀嚼无力、吞咽困难等, 患者余生非常痛苦。
7.3. 关于泰它西普的患者援助策略
为了增加SLE患者对泰它西普的可及性,荣昌推出一系列优惠计划:➡ “爱早享”购药优惠 在新药到货前患者仅需支付199元,即可获得总价值4000元理赔福利券(500元×8张),每位患者仅有一次购买机会,每张券仅限使用一支泰它西普。➡ 患者援助计划 援助对象为符合医学条件的低保和低收入的患者在临床使用16支泰它西普药品治疗后,根据检查结果,并经项目医院医生评估,确需泰它西普进一步治疗,并经项目办公室审核通过后,可凭医生处方领取32支援助药品。项目执行期间,患者可以按照此方案多次申请,当项目医生认为患者不适合继续使用泰它西普或患者自愿退出时,项目终止。
本文来自微信公众号“国广有话说”,智通财经编辑:玉景。