

本文来自 微信公众号“Insight数据库”,作者:微微阳
9 月 25 日,基石药业(02616)合作伙伴 Blueprint Medicines 宣布,近日欧盟委员会(EC)有已有条件批准
AYVAKYT®(阿泊替尼)上市销售许可,单药治疗携带血小板衍生生长因子受体α (PDGFRA) D842V 突变的不可切除性或转移性胃肠道间质瘤 (GIST)
成人患者。

本次获 EC 批准是基于基于 NAVIGATOR 一期临床试验的疗效、安全性资料,以及 VOYAGER 三期临床试验安全性结果。临床结果显示,在先前接受过或未接受过治疗的 PDGFRA D842V 突变 GIST 患者中,avapritinib 治疗表现出深度、持久的临床应答。
在 38 例 PDGFRA D842V 突变 GIST 患者中,初始剂量为 300 mg 或 400 mg,每日一次,Ayvakyt 治疗的总缓解率(ORR)为 95%,其中 13% 的患者达到完全缓解,中位缓解持续时间(DOR)为 22.1 个月,中位无进展生存期(PFS)为 24 个月,中位总生存期(OS)尚未达到。
临床中最常见的不良反应(≥20%)是恶心、疲劳、贫血、眶周水肿、面部水肿、高胆红素血症、腹泻、呕吐、周围水肿、流泪增多、食欲下降和记忆障碍。
GIST 是基因驱动的胃肠道(GI)肉瘤,大多数患者的诊断年龄在 50 至 80 岁之间,数据显示大约 5% 至 6% 的原发 GIST 病例是由 PDGFRA D842V 突变引起的,PDGFRA D842V 突变是最常见的 PDGFRA 外显子 18 突变。已发表的数据显示,用伊马替尼和其他批准的疗法治疗的 PDGFRA D842V 突变体 GIST 患者的预后较差,包括中位 OS 为 15 个月,中位 PFS 为 3 个月和 ORR 为 0%。
值得注意的是,该药是欧盟批准的第一个针对携带 PDGFRA D842V 突变 GIST 患者的靶向疗法。2020 年 1 月,avapritinib 已获 FDA 批准用于治疗携带 PDGFRA 基因 18 号外显子突变(包括 PDGFRA D842V 突变)的不可切除性或转移性 GIST 成人患者,商品名为 Ayvakit。
2018 年 6 月,基石药业与 Blueprint Medicines 签订了独家合作和许可协议,以在中国大陆,香港,澳门和台湾开发和商业化 avapritinib。
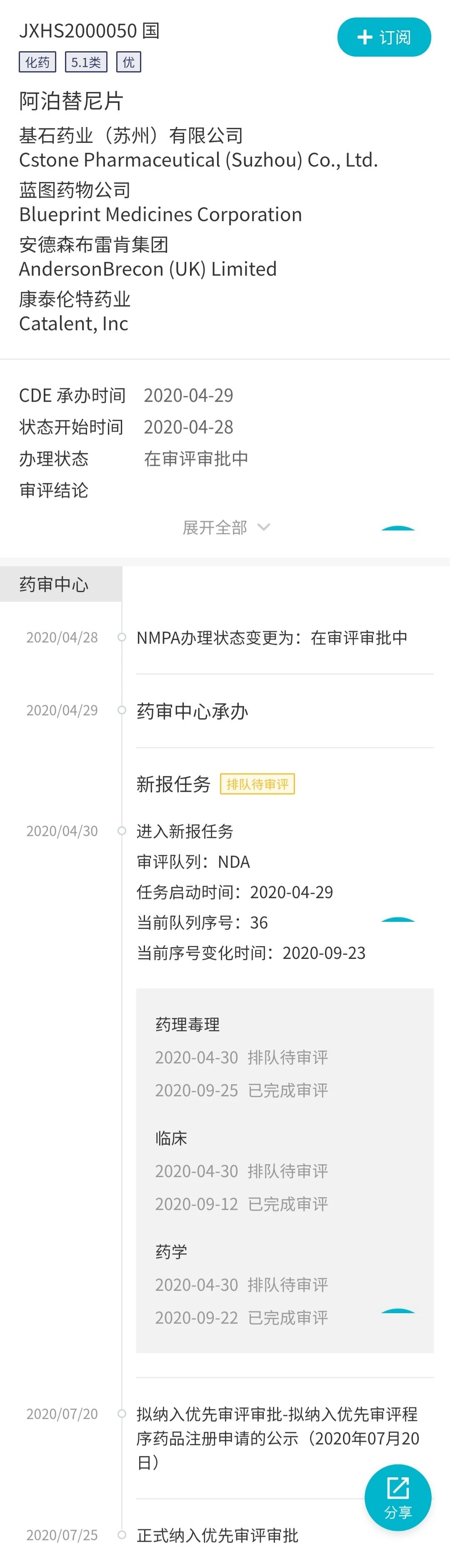
在国内,基石药业已于今年 4 月 29 日在递交阿泊替尼片的上市申请(JXHS2000050),Insight 医药情报助手显示,目前已被正式纳入优先审评,纳入理由是符合附条件批准。
在近期举办的 2020 年CSCO 年会上,基石药业公布了阿泊替尼片(Avapritinib)在中国 I/II 期桥接研究取得的积极结果。
此项桥接研究是一项开放标签、多中心的 I/II 期临床研究,旨在评估阿泊替尼治疗不可切除或转移性晚期 GIST 患者的安全性、药代动力学特征和抗肿瘤疗效。依据 I 期剂量爬坡研究的初步结果确定 II 期临床研究的推荐剂量(RP2D)。
截至数据截止日期 2020 年 3 月 31 日,共计 50 例中国患者纳入阿泊替尼的安全性评估,8 例携带 D842V 突变的患者以及 23 例 4L+患者疗效可评估。I 期研究中,患者在 200 mg 和 300 mg 剂量下对该药均显示出了良好的耐受性,研究中未观察到剂量限制性毒性。在中国 GIST 患者中的 II 期研究推荐剂量确定为 300 mg,每日一次口服,这与针对晚期 GIST 全球研究 NAVIGATOR I 期研究中的数据保持一致。
阿泊替尼在携带 PDGFRA D842V 突变的患者中初步显示出了显著的抗肿瘤活性。在 300 mg 每日一次的剂量下,8 例携带 PDGFRA D842V 突变的患者中,所有患者靶病灶均有缩小,有 5 例患者达到了研究者评估的部分缓解,总体缓解率(ORR)为 62.5%,另 3 例患者的研究者评估结果为疾病稳定。阿泊替尼在至少接受过 3 线既往治疗的(4L+)GIST 患者中也显示出一定的疗效,研究者评估的 ORR 为 26.1%。
安全性方面,阿泊替尼总体耐受性良好,研究中报告的治疗相关不良事件(TRAE)大部分为 1 级或 2 级。最常见的治疗相关 TRAE 为贫血和血胆红素升高。最常报告的≥3 级(均为 3 级)TRAE 为贫血。
编辑:(庄东骐)




